首发于 生信情报站
切换模式

生物信息之多序列比对,进化树分析,保守位点分析

白墨 
芝加哥大学 生物学系博士后
# 序列下载与整理
网址: https://www.ncbi.nlm.nih.gov/gene
下载fasta格式序列
- 输入你想查找的序列,比如Syp基因
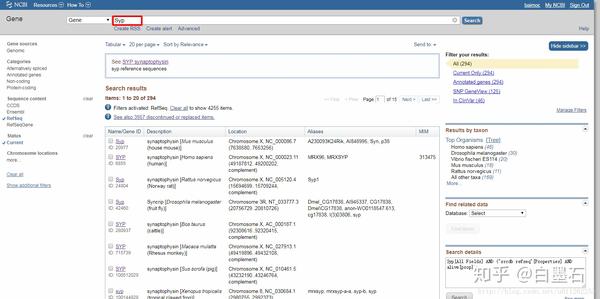

- 进入基因详细信息页面
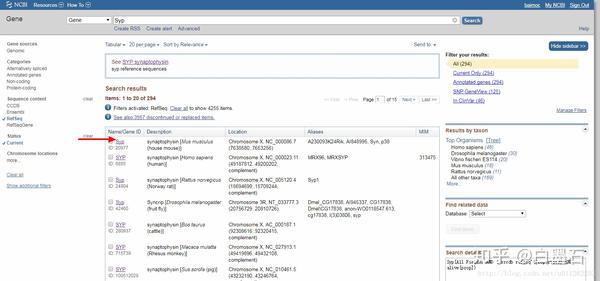

- 点击Genbank
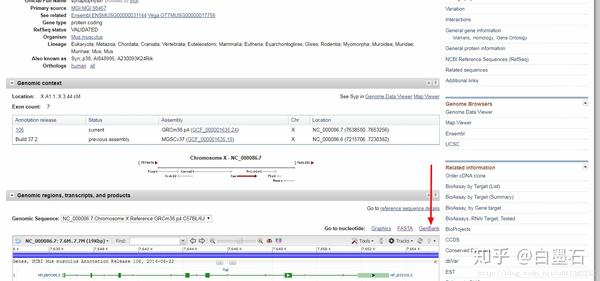

- 如图所示可以下载到fasta格式的序列,注意这里下载的是基因或者蛋白质的全序列


- 假如你希望得到promoter的基因,可以在如图所示的位置输入起始位点和终止位点
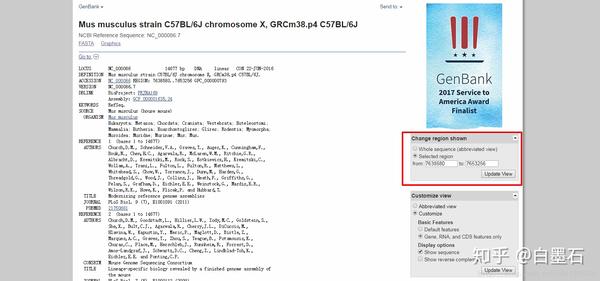
- 一般promoter的位点不确定,可以通过将起始位点左右2kb基因视为promoter
- 比如:如图起始位点为7638580,那么起始位点要减500,终止位点加1499,这时需要在from输入7638080,to输入7640079(得到长度为2kb的序列)
- 点击
Update view按钮 - 然后和同上一步下载fasta序列

合并多个fasta文件
- 下载多个序列后,你的文件夹应该是这样的
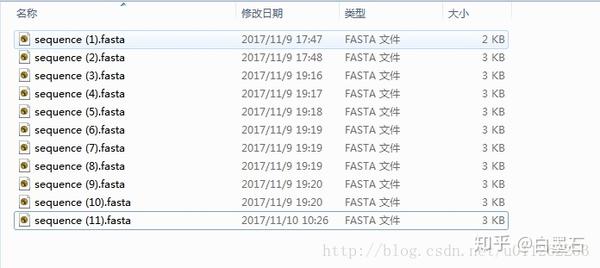

- 在文件夹空白地方
Shift+右键,点击在此处打开命令窗口,或 PowerShell
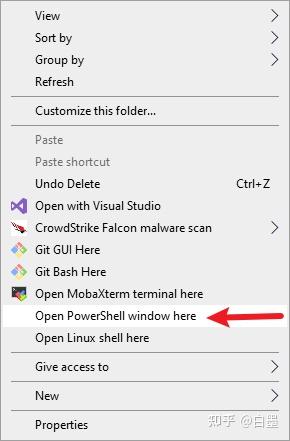
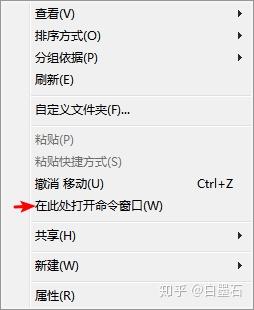
- 输入
type *.fasta > ../all_sequence.fasta
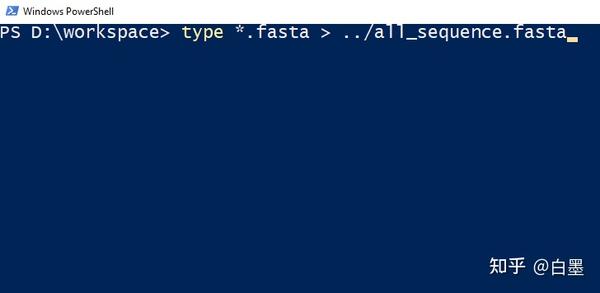

为了避免有的小伙伴在多次输出该命名造成重复的 fasta 记录,我们改为将其生成在上级目录
- 你会在上一层目录中,得到整合文件 all_sequence.fasta(这个文件也可以通过记事本打开,下面软件为UE)


多序列比对
Clustalw,Clustalx 与 MEGA的下载安装
Clustalw 下载链接: http://www.clustal.org/download/current/clustalw-2.1-win.msi
Clustalx 下载链接: http://www.clustal.org/download/current/clustalx-2.1-win.msi
MEGA 下载链接: http://www.megasoftware.net/releases/MEGA7.0.26_win64_setup.exe
序列比对
- 打开MEGA,进入序列比对分析
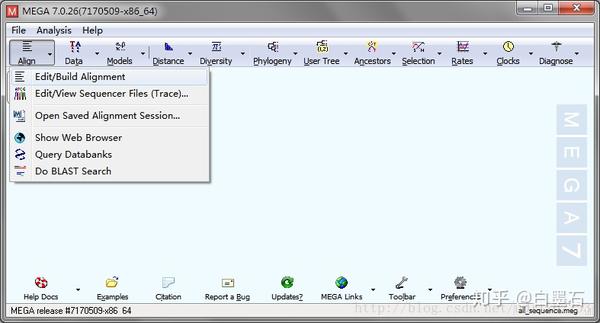

- 载入fasta序列
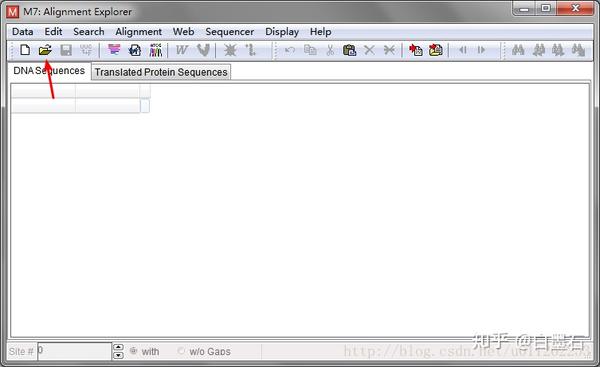

- 使用Clustalw 比对序列,参数默认点OK


- 跑出来的结果需要编辑第一列只留下物种名,序列去掉5',3'端的空序列(因为要比对序列同源性,最好把显示
-的序列去掉,使多序列的两端整齐,类似矩阵)


- 导出fasta格式和MEGA格式两种格式


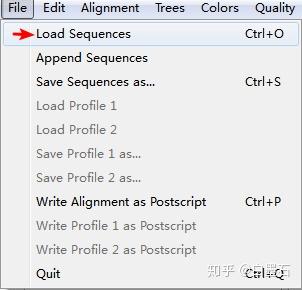
- 打开Clustalx 加载刚刚比对完的fasta格式(注意是比对完的,文件后缀名为.fas)
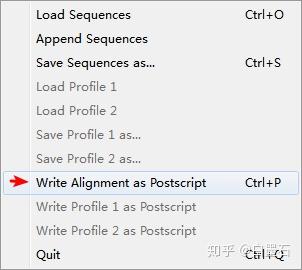
- 导出可视化文件,参数默认点OK
- 得到可视化的多序列比对结果,打开类似这样(打开用到的软件为Adobe Acrobat)


进化树分析
- 打开MEGA,载入meg文件
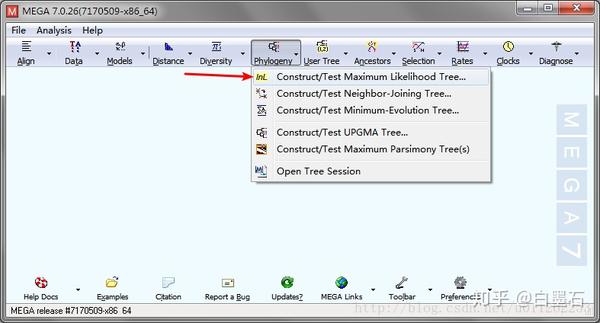

- 参数设置(这里是核酸序列)


- 得到进化树


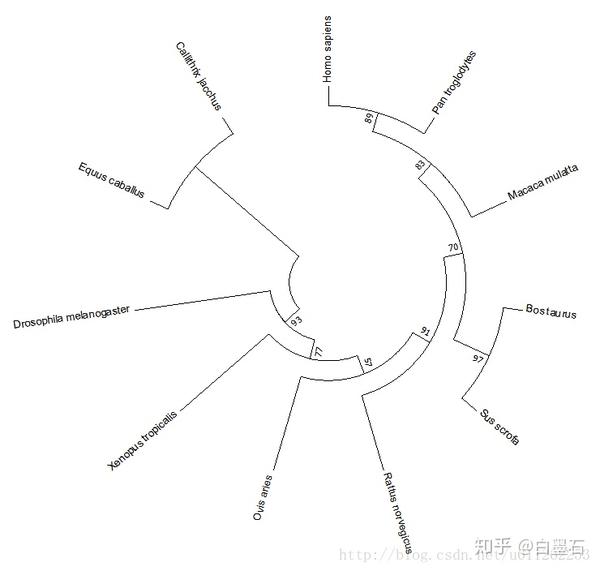

- 导出与美化


美化参考: http://www.sohu.com/a/130616941_278730
保守位点分析
- 输入网址
MEME : http://meme-suite.org/tools/meme - 上传fasta序列(这里的序列是整合后的文件,文件后缀.fasta),并输入参数(这里设置motif为10)


- 得到保守位点分析结果


编辑于 2024-04-11 01:30・IP 属地美国
生物信息学
生物学
进化
文章被以下专栏收录
