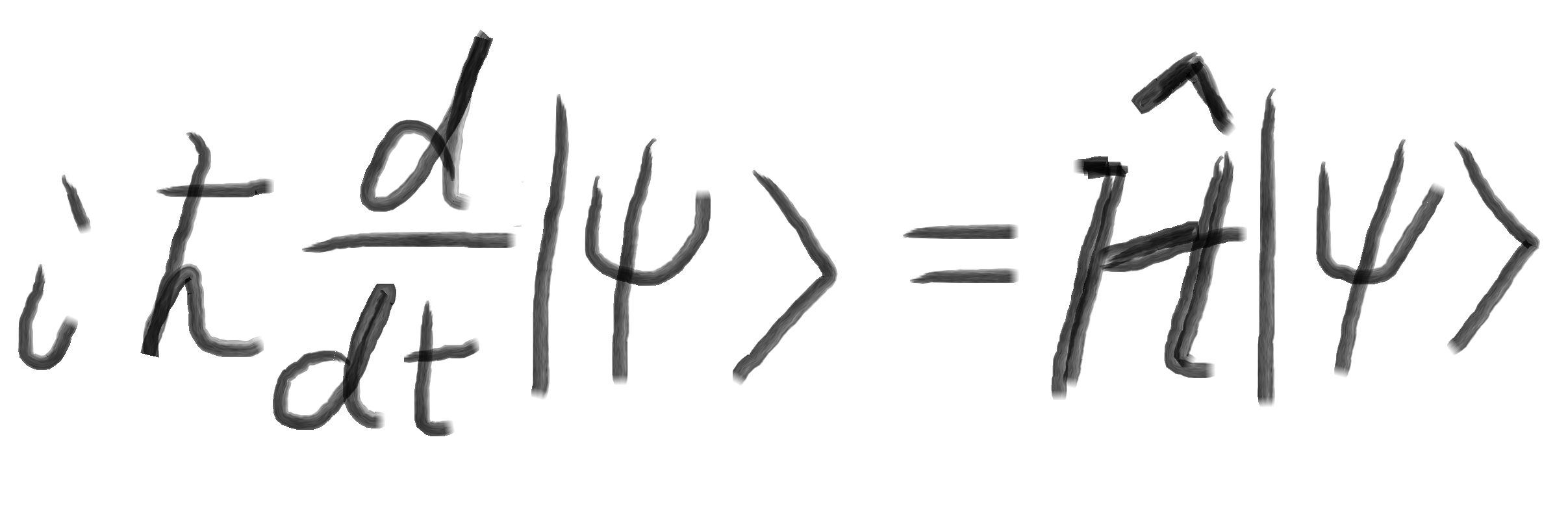
MPOC 笔记 第十四章 (8)分子轨道理论在有机化学中的应用2

继续看一些MOT在有机中应用的例子。
14.5.3 平面甲烷

物理有机化学研究的一个重要方向就是剧烈的角度弯曲及平面的四配位碳。目前应用的方法有两种,一种运用环的约束将通常的四配位的碳拗成一个平面模式,如四环壬烷类分子。如页边图所示的[4.4.4.4]Fenestrane还正在合成中。另一种方法利用了平面四配位碳的独特的电子结构,更加简便,之后再细说。注意,[4.4.4.4]Fenestrane命名不是正式的。
不要觉得书上花时间讲这种奇奇怪怪的东西莫名其妙,虽然非常小众,但一直都有人在研究,IUPAC也介绍了相关故事 [1]。下图这篇2015年的综述 [2]一上来就是1860年之前的甲烷模型,要知道von't Hoff提出碳四面体模型前,甲烷可被认为是平面的。

2020年3月的一篇文章 [3]回顾了关于下面几个分子的讨论,其中2已经被合成了。如果不是3的话,下面这图可能会把认为是行测的图推吧。
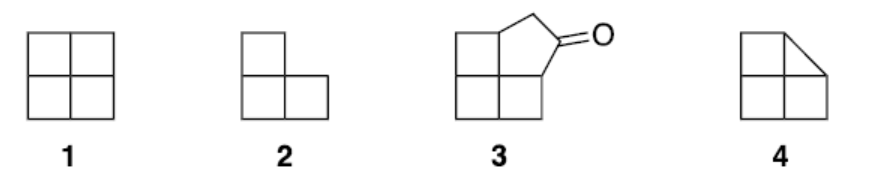

和上图3骨架类似的烷烃报道中还很稳定。


Angew和书上的图,都提到了平面碳一个代替结构,即前文的“另一种方法”二锂代环丙烷。这个结构为什么被认为很可能是平面构型的呢?来看看为什么正常的甲烷不是平面的。


先看一下平面甲烷的轨道组成。最低能量轨道来源于碳2s轨道和氢的相内组合。其次,简并轨道MO ,来源于碳的px 轨道、py 轨道和氢的原子轨道的相互作用。剩下的碳pz 轨道不能与氢的原子轨道相互作用,形成垂直平面的p轨道。最后,放入八个价电子,(四个来源于碳,每个氢提供一个)。平面甲烷含有一对占据碳的p 轨道的孤对电子。因而,只有六个价电子用于构建这四个C-H 键,形成了两个类似三中心两电子键的体系。这就解释了四面体的甲烷比平面甲烷更稳定的原因,因为一般成键越多能量越低。四面体结构的C- H 键中包含八个价电子,而平面结构只用了六个。可以回顾一下正四面体甲烷的分子轨道模型,比较一下能量差别。


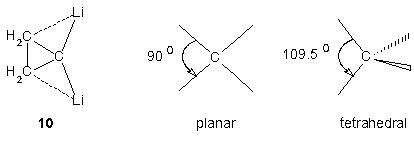
上面的认知为我们提供了一种获得稳定平面碳的方式,任何能够稳定孤对电子轨道的因素都能够稳定平面构型的碳原子。具有低能的空p/π 轨道的取代基可以作为π 受体,另一个方面相对于四面体构型的碳(109.5°) 来说,平面构型的碳的C-C 夹角较小(90°) 。考虑到这些,Schleyer 提出了一种假想的结构,如1,1-双锂环丙烷。小环倾向于小角度,且锂的空轨道能够稳定孤对电子。根据头计算研究预言,1,1-二锂环丙烷和相关的分子都倾向于这种平面构型。不过这个分子的气相数据,目前还没有。
其他有趣的结构可以看上面的综述。
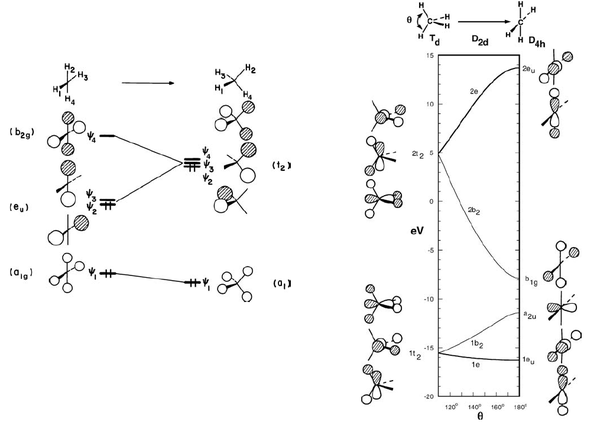
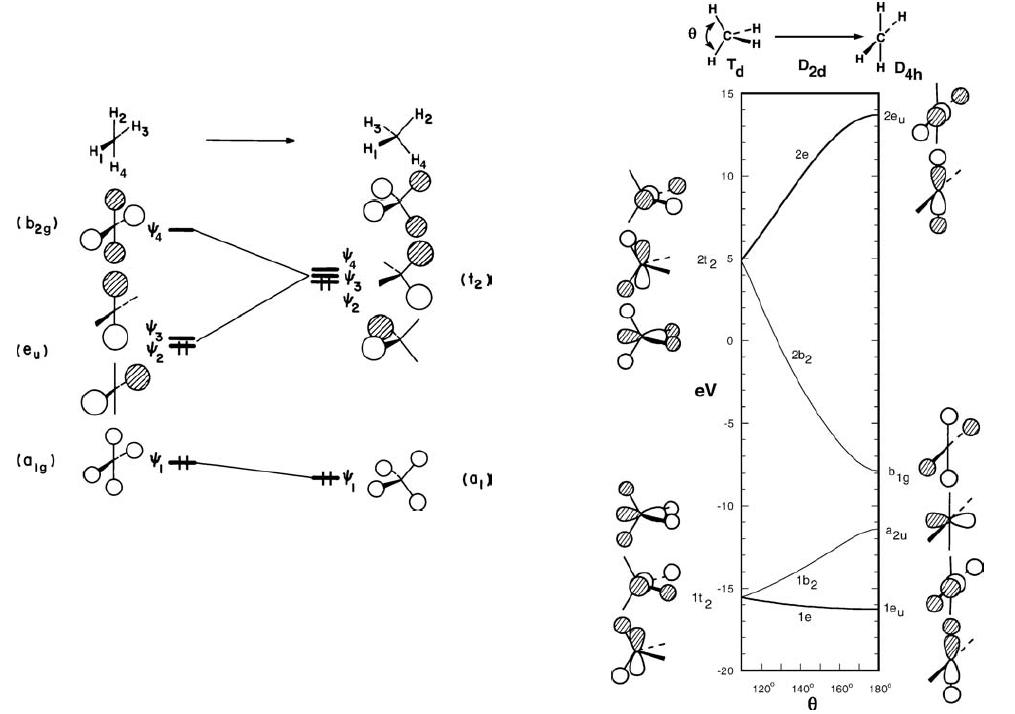
下面的Walsh图需要一些对称性的知识,不懂可以跳过。左边是H₄组成平面正方形和正四面体两种线性组合轨道的能级图,右边是甲烷中的情况,注意其中没有画a1g,左边要填满三个1t₂,右边要填到a₂u的孤对电子,明显右边平面甲烷结构的能量更高。
14.5.4 通过键的偶合Through-Bond Coupling
不知道的话看这题目可能觉得不知所云,“Through-bond”和“Through-space”是两类“超距”的相互作用 [4],经常用来解释一些共轭体系之间能量传递的现象,现在的MOF、COF等体系中研究的很多。“通过键耦合”这个翻译只能说凑合吧。这个概念比较新,叫法也很多,所以书上没有给出Through-Bond Coupling到底是什么下面是一些综述中的说法。
Through bond interaction, called variably spin-spin coupling, J-coupling, scalar coupling, or coherence transfer, occurs because the spin state of a proton affects the spin state of its valence electron.
这个问题可以通过考虑1,4- 环己二烯(也叫1,4 -二氢苯)来分析。问题集中在和这两个双键相连的π 轨道。首先分析π 轨道的线性组合:一个相内组合(对称,symmetric,S) 和一个相外组合(反对称,antisymmetric,A) 。当双键间没有任何直接的相互作用时, S 将有可能位于A 之下。这种直接混合称之为通过空间(through space)的偶合。多数实验结果表明,在1,4-环己二烯中轨道的实际顺序是A< S。因为这是一个平面分子,所以存在一个可以从σ 框架中分离处的π 电子体系。对于相内轨道混合则不一定合适。
P.S. 注意antisymmetric和asymmetric (不对称)是不同的概念。
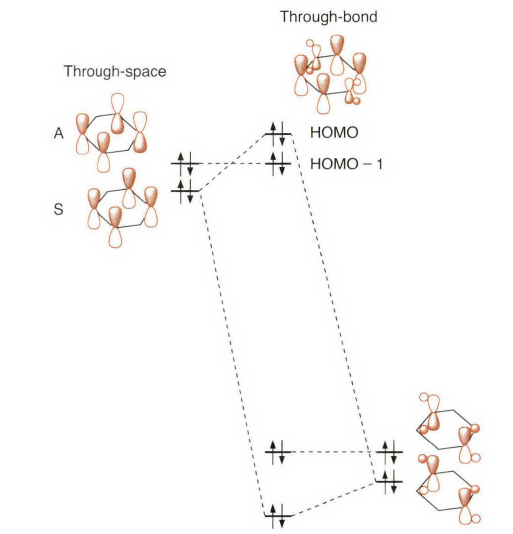
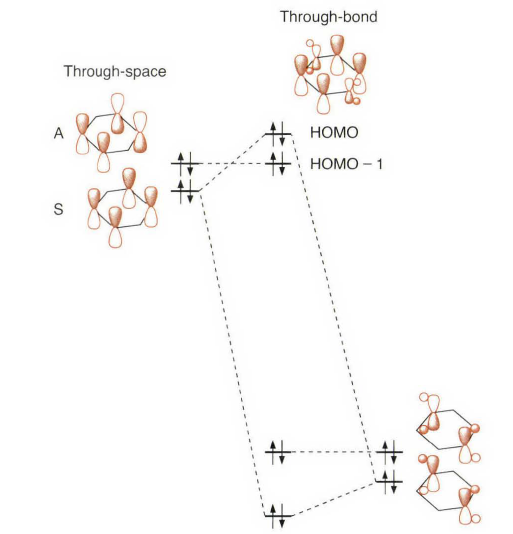
不适合的原因在于并没有考虑到所有的π 轨道。第1章后半部分的一个重点便是关于亚甲基也具有π 对称的MO 轨道,而环己二烯就是一个完美的体系。如上图所示,对π(CH₂)轨道位于传统的π 轨道与组合的A 的节面上,因此不能混合。然而, π(CH₂) 轨道可以与组合的S 混合。从而降低了π(CH₂)轨道的能量,但是提高了S 组合与π 轨道的能量。当这种相互作用足够强时,就会导致能级顺序为A<S。环己二烯的真正的分子轨道证实了这种猜测。


这和第一章中烷基基团提高了烯键π 轨道的能量是一个道理。
14.5.5 正离子的独特成键能力-非经典离子和超价键碳
本书中第一、第二和第十三章都介绍了中间体碳正离子,当时说这些中间体寿命很短,实验测定很难,多是靠量子化学结果。
简单回顾一下,第1 章中,我们讨论了碳正离子异常的成键能力,并着重指出三中心二电子的结构对形成桥连结构的重要性。对于经典的乙基正离子,它主要存在两种形式:通过强烈超共轭作用形成的传统碳鎓离子和完全桥连的碳鎓离子。第2 章中,我们就桥连作用的性质如何进一步提高碳鎓离子的稳定性进行了说明,氢亲和值的降低是明显的证据。第11 章中,我们介绍了取代反应如何经由非经典碳正离子进行,以及这一过程中碳-碳骨架发生不规则变化的有趣现象。
这个内容实在太多了,因此,我们在本节中将只考虑非经典离子结构方面的问题,尤其是基于理论和实验方法对各种离子的结构预测。首先,我们就过渡态结构的计算方法进行一下必要的介绍。


过渡态结构的计算方法
忘记的可以回顾一下动力学章节(7-9章)中的势能面的概念。如上图所示。对于物理有机化学的基本问题连续统的可能性问题,传统的情形如A 所示,在这种情况下两种碳鎓离子可经由桥连碳鎓离子过渡态相互转化。在这一体系中,对称的碳锚离子将两种不太对称的碳鎓离子联系起来。该桥连的碳鎓离子本身可能就是一种稳定的结构,相对于不太对称的碳鎓离子,它的势能面可能高(B,也可能低(C) 。还有一种情形,如D 所示,势能面上唯一稳定的结构就是对称的碳鎓离子过渡态。对于非经典离子的假设,问题在于究竟B 、C 和D 所示的势能面中哪种是可行的。
理论有机化学提供了两种理论证据。其一是用分析方法表达简正坐标下分子从头计算法能量的一阶和二阶偏导。本章开头和第二章分子力学部分中我们利用分子机理方法讨论了几何优化的问题,分子能量表达式的一阶和二阶偏导分析可以高效而完全地优化分子的几何构型;不过这里不用分子力学方法,而是转化成相应的从头计算法波函数。得益于此,近似和局部的分子几何优化成为可能。但对于非经典离子问题,这种解决方法还不能令人满意。
具体的例子见上图E,这里要看懂讨论的内容需要回顾一些东西。从头计算法几何优化最重要的方面是能够通过分析分子能量的偏导给出原子坐标,这就方便我们从过渡态中找到真正的极小值。当分子内力为零时,即分子能的一阶导数dE/dx(x 为几何坐标)为零,称分子对应的结构为稳态点,这个在第二章分子力学中讨论过。在计算中,几何坐标应该选取分子的简正坐标,这在第2 章和第7 章己讨论过。简正坐标的数目对应于分子所有可能的振动模式,对于绝大多数的有机分子有3N-6 种简正坐标,其中N 表示分子中原子的数目。
分子能量对于每一简正坐标的一阶导数,称为梯度分量,也就是斜率;因此,稳态点就对应所有斜率均为零的那点。对于分子能量二阶导数的表达式d²E/dx²,是极值的取向;当d²E/dx²为正时,是极小值;当d²E/dx²为负时,是极大值。
二阶导数,也叫本征值,只有通过它,我们才能够分辨哪种是过渡态,哪种是稳定的分子。对于稳定的分子,其所有的本征值都是正值;而对于过渡态,其本征值有且仅有一项为负值。
其二是发展了一系列从头计算法并运用于有机分子核磁位移的预测。其中应用最为广泛的当属由Kutzelnigg 和Schindler 开发的IGLO( 区域化轨道分立算法)。进一步的研究表明,通过扩展的基组和更高级的几何优化理论(如MP2) ,理论计算的核磁化学位移与观测值能够极好地吻合。这些方法的深度超出了本书的范围,所以没展开讨论。
从头计算法对碳正离子的应用
对于碳正离子化学来说,理论与实验交汇的成果之所以如此丰硕,主要有以下几个方面的原因。1)碳正离子有丰富的热力学数据,2)大量结构性数据可以在稳定离子媒介条件下通过核磁方法检测,3)所涉及的碳正离子均为相当小的分子,这为更高级理论的应用提供了便利,4)碳正离子对于给定的基组减少了一个电子,这会使相关能起到的作用更小,尽管看上去这种影响好像很小,但是在相同的理论条件下对正离子的处理的确比对负离子容易得多。
碳正离子的核磁位移
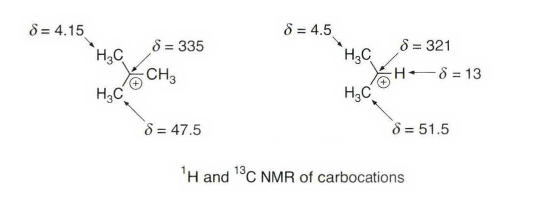

正离子中心的存在对化学位移有着显著的影响。对于带正电荷的碳原子,向低场方向有着相当大的位移,且相邻的碳原子向低场方向亦有不同程度位移。对于碳正离子,这种向低场方向的位移相当普遍,其位移值可由下式计算,其中δ 是碳原子的¹³C 化学位移。通过对应烯烃的比较,从而求得碳正离子中所有碳原子向低场方向位移的总和ΔΣ。对大量典型碳鎓离子的研究表明ΔΣ的数值相当恒定,通常为350~390 ,在一些特例中可能有更大的观测值。

我们期望三中心二电子的碳鎓离子有着相当典型的ΔΣ值;特别是对于超价键碳。实验表明:相对于己观测到的碳鎓离子而言,其存在相当大的高场位移,导致ΔΣ数值大大减小。比如下面这个,ΔΣ=-1.


这里我们讲的都是核磁内容,讲一下它和计算的关系。上图是7- 降冰片烯碳正离子,ΔΣ 数值在正常值范围之外,表明了形式上的碳正离子中心C7 与C2-C3 双键的作用非常强烈以致产生了一种新的成键方式, 三个碳原子通过两个π 电子有效地联系在一起,是典型的三中心二电子成键方式。该离子随同HOMO 和LUMO 一起的计算结构,也指出C7 是如何强烈地靠向烯碳这一侧,与采用轨道混合方法预测的结果完全吻合。通过协同和非协同的方式组合了烯π 键和C7 的空p 轨道, C2 -C3 双键比典型的双键键长要长,这应该是由于π 电子密度分布由C2-C3 成键区向C7 转移的结果。
如果一个适当方位的烯键与碳正离子中心相互作用生成碳鎓离子,那么也可以在环丙烷环结构中得出相似的结果。另一个碳正离子的例子源于五环壬烷,也叫Coates 离子,即使是最激烈的非经典离子概念的反对者也认同这是一个桥连对称的碳偷离子,ΔΣ数值也完全支持了这一解释。
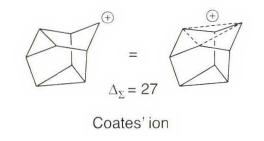
与碳鎓离子相关的特殊化学位移也存在于¹H NMR 中。看下面这个例子。

降冰片基碳正离子
这个分子的性质有多神奇可以看第11章。
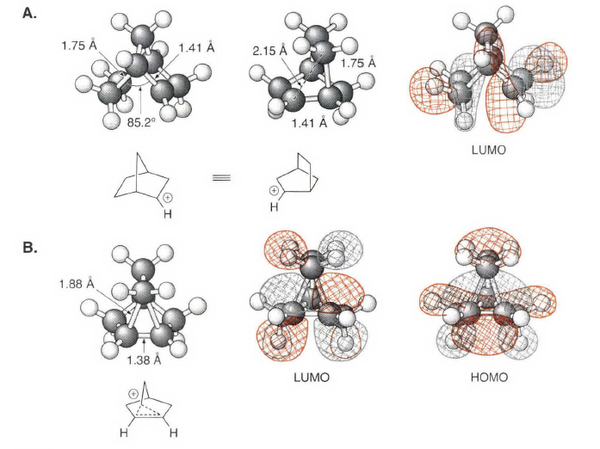

2 -降冰片基碳正离子既表现出经典碳鎓离子的特性,又表现出非经典碳鎓离子的特性。如果假设它具有碳鎓离子的经典结构,那么这个体系就容易在取代反应中发生重排。尽管,非经典的桥连结构已可以解释该取代反应中的众多产物,通过量化计算得到的如图所示的结构却能够更好地说明其内在的细节,这仅仅在于CH₂基团是否靠右侧。尤其对于降冰片基碳正离子来说,这一细节的不同关系到非经典离子的核心问题。
核磁数据也支持了这一桥连的结构。在稳定离子条件下,其ΔΣ的数值为175 ,尽管没有Coates 离子与典型碳鎓离子间数值那样巨大的差别,这一数值也比典型碳鎓离子的数值范围低很多。核磁数据支持了桥连碳正离子的结果。
14.5.6 自旋取向
本章前面的章节中,在讨论卡宾时介绍过单线态和三线态卡宾的不同,更多的例子,比如双自由基化合物、单线态氧和高自旋化合物之类的后面的章节会介绍。下面讨论氢分子和碳原子。
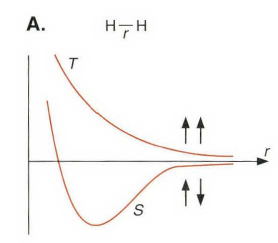
就键长而言,基态的氢分子是单重态,三重态是能量很高的激发态。为了在氢分子中成键,必须将这两个共用电子放到成键分子轨道中,这两个电子成对且符合Pauli 原理。而在三重态中,一个电子在成键分子轨道,另一个电子在反键分子轨道,不存在H-H键。尽管高级的量化计算一致给出没有任何三重态的势能面会在单重态之下的结果,但是如果拉伸H-H键,成键和反键分子轨道之间的能隙便会消失,同时其单重态和三重态也将同一化。
如果缺少任何特定的影响,那么具有弱相互作用双电子的体系将会是单重态,原因是有较少的成键作用也比没有任何作用好。如果两个自由基可以相互靠近而且轨道重叠,无论它们是同一分子(双自由基) ,还是两个相互靠近的单自由基,元论是在搭液状态中还是在晶格条件下,最可能的结果就是生成低自旋的单重态分子。
碳原子有四个价电子,其中两个成对电子占据碳原子的2s 轨道,另外两个碳原子占据三重简并的2p 轨道, p 轨道中的两个电子高自旋配对,碳原子呈现基态三重态。下面这个图在杂化理论里经常出现,现在想想是不是很奇怪?

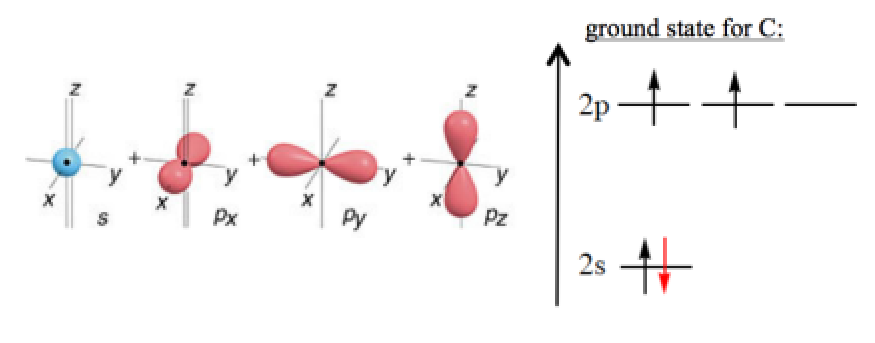
碳原子和氢分子不同是因为碳原子的情形下交换积分起到了关键的作用,由于轨道的波节性,碳原子两个未成对电子所占据的p 轨道彼此正交,当然,这并不意味着两电子之间完全没有相互作用,但是,这两个正交p 轨道的重叠积分为零。重叠意味着成键,但是成键却需要低自旋态,因此高自旋态的首要条件就是轨道重叠积分为零或近似于零。而拉长键氢分子的情形,尽管非常弱的轨道重叠积分也可以产生单重态取向,但是由于两个氢原子离得实在太远,原子轨道的重叠积分也就太小。

如上图所示,尽管碳原子原子轨道的正、负重叠区为零,这两个轨道实际上通过部分占用相同空间相互作用,也可以称为协同扩展作用,净相互作用仍然为零。这就是同中心原子轨道令人难以理解的特性一一它们在相同空间里有着极强的协同扩展作用,原因在于交换积分K 并非像轨道重叠积分那样遵循相同的波节性。
由于没有迹象表明对轨道重叠积分作平方处理会有什么差异,因此原子轨道正、负区域的重叠并不意味着它们能够相互抵消,为了更好地近似这一情形,我们将交换积分的数值用轨道重叠积分的平方表述。同样的,碳原子轨道重叠积分为零,交换积分却并非如此。我们讨论过,交换积分只有在自旋平行时才能起到稳定化的作用;如果没有成键机会,或者说没有自旋配对的强烈驱动力,那么高自旋态将是更好的稳定取向,这就是Hund 规则的由来。因此,在这种情形下,碳原子更倾向于高自旋态,表现为基态三重态。对于长键氢分子,当拉伸H-H键时,其轨道重叠积分和交换积分都相应减小,但由于轨道重叠在任何键长下都能发生作用,于是其表现为基态单重态。
后面的很多地方会用“光谱项”却没有介绍,应该是默认都懂吧。这里说一下,对于给定的原子组态,如果确定了角量子数L和自旋量子数S,会写成 ^{2S+1}L 。
对于原子谱相L=0,1,2,3……时,L写成S、P、D、F……
ns²因为Pauli 原理,只能是¹S;而1s¹2s¹可以单线态可以三线态,可能是¹S,也可能是³S。
对于给定的分子组态,如果确定了总角量子数 M_L 和自旋量子数S,会写成 ^{2S+1}\Lambda 。
对于原子谱相M_L=0,1,2,3……时, \Lambda 写成Σ、Π、Δ、Φ……
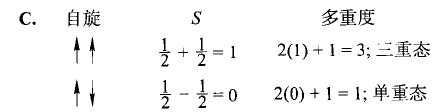

分子中的三线态基态最有名的就是氧气分子,单线态氧的问题会在后面光化学的最后一节讲。
讲两个有机分子,环丁二烯(CBD) 和三亚甲基甲烷(TMM) 。
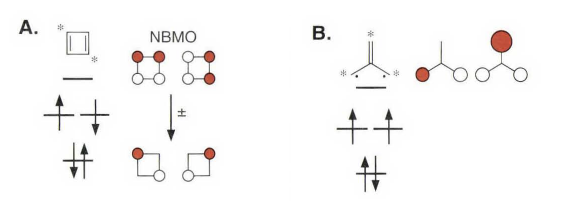
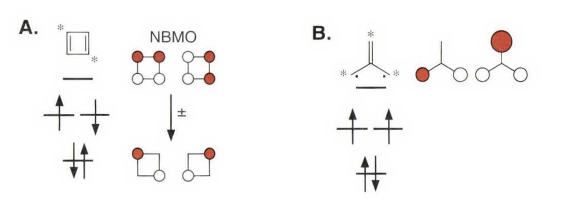
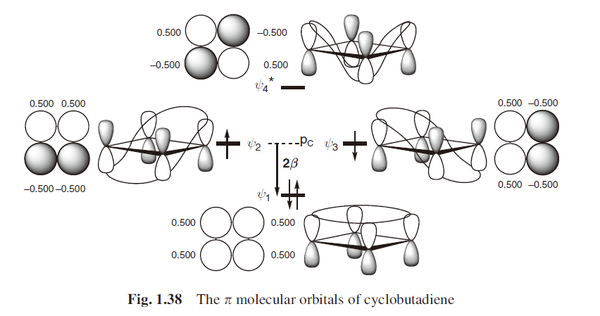
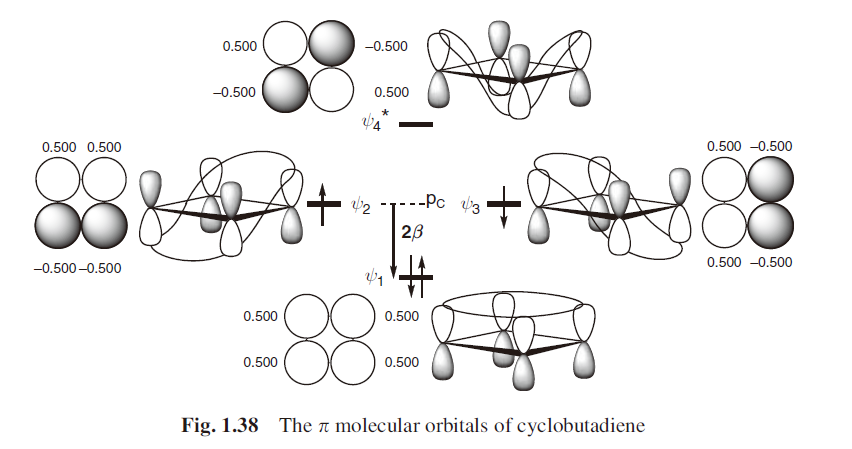
环丁二烯和三亚甲基甲烷具有数目相同的分子轨道,它们都有一对简并的非成键分子轨道(NBMOs),并由两个电子占据。但实际上这两个体系的自旋不同,环丁二烯是单线态和三亚甲基甲烷是三线态,取向的能量量值却是在同一量级,对于单重态的正方形环丁二烯,其数值约为10 kcal/mol;而对于三重态的三亚甲基甲烷,数值约为14 kcal/mol。(图上还标出了交替碳氢化合物的星号)
考虑这两个体系非成键分子轨道的区别,就能解释它们自旋取向不同的原因。对于一对简并的分子轨道,可以进行线性组合。对于环丁二烯体系,可以得到没有相同原子的线性组合,一个非成键分子轨道被限定在1 位和3 位碳原子上,而另一个非成键分子轨道被限定在2 位和4 位碳原子上,它们分别占据两组不同的碳原子,这种轨道的线性组合称为非交集组合。与此不同,在三亚甲基甲烷体系中,无论怎样组合,都不能找到非交集的非成键分子轨道线性组合,不同组中总有相同的原子,那么这种非成键分子轨道的线性组合就称为交集组合。三亚甲基甲烷类似的体系有六个电子时有时也叫“Y芳香性”,包括一些和碳酸根,胍类似的六电子体系。
类似讨论轨道的交换和重叠那样,体系中非成键分子轨道是否存在交集非常重要,因为无论在环丁二烯还是三亚甲基甲烷中,非成键分子轨道都是正交的。环丁二烯体系中,非成键分子轨道非交集组合,轨道交换积分也相当趋近于零。这就好像一个电子被限定在1 位和3 位碳原子上,而另一个电子被限定在2 位和4 位碳原子上一样,其两个非成键电子在空间上不能协同扩展。因此,无论是对于第一规则(单电子理论) ,还是第二规则(包括电子排斥理论) ,单重态和三重态都是简并的。
不过现实中环丁二烯是单线态的,还和伪Jahn - Teller 扭曲有关。可以发现环丁二烯的结构并不是正方形,而是长短键交替变化的长方形,其中长键键长为1. 54埃,短键键长为1. 37埃。看上去它更像一个双烯分子,而并非两种相同形式的烯烃的共振互变混合体。
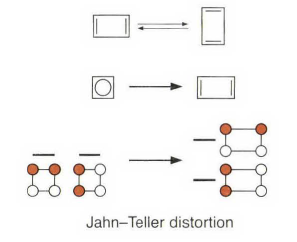
这里要注明一下,书上环丁二烯的分子轨道图画错了,画成了三线态,所以看起来有些莫名其妙。更多的讨论可以参考其他书籍。


上一节:
下一节:对称性。
目录:
- 《现代物理有机化学》笔记 第一章 化学键基础(1)
- 《现代物理有机化学》笔记 第二章 张力和稳定性(1)
- 《现代物理有机化学》笔记 第三章 溶液和非共价结合力(1)
- 《现代物理有机化学》笔记 第五章 酸碱(1)
- 《现代物理有机化学》笔记 第六章 立体化学(1)
- 《现代物理有机化学》笔记 第七章 动力学(1)
- 《现代物理有机化学》笔记 第八章 机理相关实验(1)
- 《现代物理有机化学》笔记 第九章 催化(1)
- 《现代物理有机化学》笔记 第十章 机理(1)
- 《现代物理有机化学》笔记 第十一章 机理2(1)
- 《现代物理有机化学》笔记 第十二章 金属有机(1)
- 《现代物理有机化学》笔记 第十四章 电子结构理论新概念(1)
- 《现代物理有机化学》笔记 附录五
参考
- ^ http://publications.iupac.org/pac-2007/1998/pdf/7010x1977.pdf
- ^http://dx.doi.org/10.1002/anie.201410407
- ^10.1021/acs.joc.0c00187
- ^ https://www.sciencedirect.com/topics/chemistry/through-bond-interaction
文章被以下专栏收录
