1,3-环己二酮的制备方法与流程
1.本发明涉及有机合成领域,具体涉及一种1,3-环己二酮的制备方法。
背景技术:
2.1,3-环己二酮作为中间体用于有机合成,可以用于材料单体、固化剂、 溶剂等多种有机化合物的合成,是除草剂磺草酮、硝磺草酮的中间体。
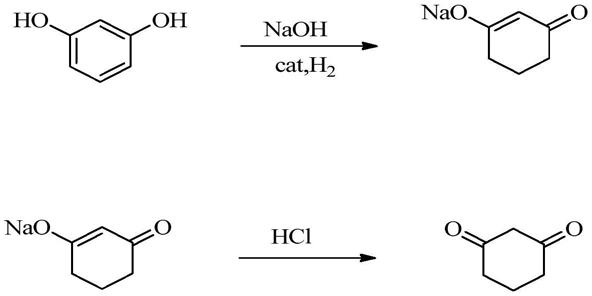
3.现有的1,3-环己二酮制备方法中,通常采用以间苯二酚为起始原料,经 加氢还原,酸化,重结晶得到1,3-环己二酮产品,其制备路线如下。
[0004][0005]
现有技术中均未有关于稳定晶型的1,3-环己二酮的报道,例如 cn200510489576.x、cn201110419160.6、jp0413644以及jp20013421632001 均是通过催化剂的选择,其对产物选择性及收率影响较大。
[0006]
因此,急需提供一种纯度高、收率高、晶型稳定的1,3-环己二酮的制备 方法。
技术实现要素:
[0007]
本发明的目的是为了克服现有技术的存在1,3-环己二酮反应液制备过程 中,1,3-环己二酮的粒径小、纯度低且晶型不稳定等的问题,提供一种1,3
-ꢀ
环己二酮的制备方法,该制备方法具有制备得到的1,3-环己二酮的纯度高、 收率高、晶型稳定且保存稳定性高等优点。
[0008]
为了实现上述目的,本发明提供一种1,3-环己二酮的制备方法,该方法 包括:以间苯二酚为原料,经加氢还原、酸化结晶的步骤,其中,所述酸化 结晶包括:
[0009]
a)使加氢还原产物与第一酸进行接触并使得接触后产物的ph为4-7后, 加入晶种,并在0-25℃下保持1-3h;
[0010]
b)通过第二酸调节步骤a)得到的产物的ph为1-3后,并在5-15℃下 保持0.1-2h。
[0011]
优选地,步骤a)中,使加氢还原产物与第一酸进行接触并使得接触后 产物的ph为4-6后,加入晶种,并在5-15℃下保持1.5-2.5h。
[0012]
优选地,步骤b)中,通过第二酸调节步骤a)得到的产物的ph为1.5-2.5, 并在5-10℃下保持0.5-1.5h。
[0013]
优选地,所述第一酸为盐酸、硫酸和硝酸中的一种或多种。
[0014]
优选地,所述第一酸为盐酸和/或硫酸。
[0015]
优选地,所述第一酸的浓度为0.5-50wt%。
[0016]
优选地,所述第二酸为盐酸、硫酸和硝酸中的一种或多种。
[0017]
优选地,所述第二酸为盐酸和/或硫酸。
[0018]
优选地,所述第二酸的浓度为0.5-50wt%。
[0019]
优选地,所述第一酸与所述第二酸相同。
[0020]
优选地,所述方法还包括将酸化结晶产物进行固液分离、干燥的步骤。
[0021]
优选地,所述加氢还原的条件包括:压力为0.5-10mpa,温度为50-150℃。
[0022]
通过上述技术方案,利用本发明的技术方案制备的1.3-环己二酮纯度大 于98.5%,收率大于90%,产品成结晶状,且在保存和运输过程稳定。
附图说明
[0023]
图1为本发明实施例1制得的1.3-环己二酮的粉末x射线衍射图谱。
[0024]
具体实施方式
[0025]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这 些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各 个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点 值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视 为在本文中具体公开。
[0026]
本发明提供一种1,3-环己二酮的制备方法,该方法包括:以间苯二酚为 原料,经加氢还原、酸化结晶的步骤,其中,所述酸化结晶包括:
[0027]
a)使加氢还原产物与第一酸进行接触并使得接触后产物的ph为4-7后, 加入晶种,并在0-25℃下保持1-3h;
[0028]
b)通过第二酸调节步骤a)得到的产物的ph为1-3后,并在5-15℃下 保持0.1-2h。
[0029]
在本发明中,通过控制酸化结晶程,大幅提高了制备的收率,且回收得 到的产品成结晶状、颗粒大且纯度高。
[0030]
对于上述以间苯二酚为原料,经加氢还原、酸化结晶制备1,3-环己二酮 的方法,其反应路线如下:
[0031][0032]
作为上述加氢还原的具体方法,可以使用现有能够完成间苯二酚加氢的 任意方法,可以将间苯二酚与无机强碱先中和成盐,再在催化剂的存在进行 加氢还原。例如可以将物质的量比为1∶1-1.5的间苯二酚与氢氧化钠中和, 然后在加氢催化剂下,在0.5-10mpa、50-150℃下进行加氢。
[0033]
所述加氢催化剂可以为本领域通常用于加氢还原的各种催化剂,例如可 以为钯炭或雷尼镍催化剂。
[0034]
根据本发明,在加氢还原后,本发明将加氢还原产物进行本发明特定的 酸化结晶
步骤,所述酸化结晶包括:
[0035]
a)使加氢还原产物与第一酸进行接触并使得接触后产物的ph为4-7后, 加入晶种,并在0-25℃下保持1-3h;
[0036]
b)通过第二酸调节步骤a)得到的产物的ph为1-3后,并在5-15℃下 保持0.1-2h。
[0037]
为了进一步促进提高制备的收率及制得的产品纯度,在本发明中,优选 地,步骤a)中,使加氢还原产物与第一酸进行接触并使得接触后产物的ph 为4-6后,加入晶种,并在5-15℃下保持1.5-2.5h;更优选地,步骤a)中, 使加氢还原产物与第一酸进行接触并使得接触后产物的ph为4.2-5.8后,加 入晶种,并在5-15℃下保持1.5-2.5h。
[0038]
为了进一步促进提高制备的收率及制得的产品纯度,在本发明中,优选 地,步骤b)中,通过第二酸调节步骤a)得到的产物的ph为1.5-2.5,并在 5-10℃下保持0.5-1.5h。
[0039]
对于上述酸化结晶过程中采用的酸,没有特别的限定,在本发明中,优 选地,所述第一酸为盐酸、硫酸和硝酸中的一种或多种;更优选地,所述第 一酸为盐酸和/或硫酸。在本发明的一个具体实施方式中,所述第一酸为盐酸。
[0040]
对于上述第一酸的浓度没有特别的限定,在本发明中,优选地,所述第 一酸的浓度为0.5-50wt%;更优选地,所述第一酸的浓度为5-20wt%。在本 发明的一个具体实施方式中,第一酸采用浓度为5wt%的盐酸;在本发明的 另一个具体实施方式中,第一酸采用浓度为10wt%的盐酸;在本发明的另一 个具体实施方式中,第一酸采用浓度为15wt%的盐酸。
[0041]
对于上述第二酸的浓度没有特别的限定,在本发明中,优选地,所述第 二酸的浓度为0.5-50wt%;更优选地,所述第二酸的浓度为5-20wt%。
[0042]
为了进一步方便操作,在本发明中,优选地,所述第一酸与第二酸相同。 在本发明的一个具体实施方式中,第一酸与第二酸同样采用浓度为5wt%的 盐酸;在本发明的另一个具体实施方式中,第一酸与第二酸同样采用浓度为 10wt%的盐酸;在本发明的另一个具体实施方式中,第一酸与第二酸同样采 用浓度为15wt%的盐酸。
[0043]
上述加氢还原产物中1,3-环己二酮的含量可以为0.5-50重量%,优选为 20-40重量%。通过使用上述1,3-环己二酮浓度含量的加氢还原产物,可以进 一步提高制得的1,3-环己二酮的纯度和收率。在本发明的一个具体实施方式 中,采用1,3-环己二酮的含量为30wt%的加氢还原产物。
[0044]
对于上述晶种的加入量,例如可以为:相对于100g的加氢还原产物, 晶种的加入量为0.1-5g,优选为0.1-1g,更优选为0.4-0.6g。在本发明的一个 具体实施方式中,在100g的加氢还原产物中,加入0.5g的晶种。
[0045]
在本发明中,所述方法还包括将酸化结晶产物进行固液分离、干燥的步 骤。
[0046]
上述固液分离可以采用静置分液,过滤或者离心分离方式进行,能够实 现分离即可。在本发明的一个具体实施方式中,将酸化结晶产物过滤并烘干, 得到1,3-环己二酮产品。
[0047]
以下将通过实施例对本发明进行详细描述,但本发明并不仅限于下述实 施例。
[0048]
以下实施例中,“收率”指的是得到的1,3-环己二酮相对于经加氢还原 得到的产物中的式所示的化合物的摩尔收率。
[0049]
实施例1
[0050]
(1)将浓度为5wt%的盐酸400g(0.548mol),降温至5℃;
[0051]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为4.5,停止滴加, 加入1g晶种,控制温度保持在5℃下搅拌2小时后,继续滴加盐酸至加氢还 原产物的ph为2,在5℃下保温1小时,将酸化结晶产物经过滤、烘干,得 到产品,其中,1,3-环己二酮的含量为99.3%,收率为90.2%,制得的1,3
-ꢀ
环己二酮的晶型的xrd谱图如图1所示。
[0052]
实施例2
[0053]
(1)将浓度为10wt%的盐酸200g(0.548mol),降温至10℃;
[0054]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为5,停止滴加, 加入1g晶种,控制温度保持在10℃下搅拌2小时后,继续滴加盐酸至加氢 还原产物的ph为2,在10℃下保温1小时,将酸化结晶产物过滤、烘干, 得到产品,其中,1,3-环己二酮的含量为98.8%,收率为90.57%。
[0055]
实施例3
[0056]
(1)将浓度为15wt%的盐酸133.3g(0.548mol),降温至15℃;
[0057]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为5.5,停止滴加, 加入1g晶种,控制温度保持在15℃下搅拌2小时后,继续滴加盐酸至加氢 还原产物的ph为2,在15℃下保温1小时,将酸化结晶产物过滤、烘干, 得到产品,其中,1,3-环己二酮的含量为98.5%,收率为90.62%。
[0058]
实施例4
[0059]
(1)将浓度为5wt%的盐酸400g(0.548mol),降温至5℃;
[0060]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为4.2,停止滴加, 加入1g晶种,控制温度保持在5℃下搅拌2小时后,继续滴加盐酸至加氢还 原产物的ph为2,在5℃下保温1小时,将酸化结晶产物过滤、烘干,得到 产品,其中,1,3-环己二酮的含量为95.2%,收率为90.1%。
[0061]
实施例5
[0062]
(1)将浓度为5wt%的盐酸400g(0.548mol),降温至5℃;
[0063]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为4.7,停止滴加, 加入1g晶种,控制温度保持在5℃下搅拌2小时后,继续滴加盐酸至加氢还 原产物的ph为2,在5℃下保温1小时,将酸化结晶产物过滤、烘干,得到 产品,其中,1,3-环己二酮的含量为94.3%,收率为90.5%。
[0064]
实施例6
[0065]
(1)室温(25℃)下,配制浓度为5wt%的盐酸400g(0.548mol);
[0066]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为4.5,停止滴加, 加入1g晶种,控制温度保持在25℃下搅拌2小时后,继续滴加盐酸至加氢 还原产物的ph为2,在25℃下保温1小时,将酸化结晶产物过滤、烘干, 得到产品,其中,1,3-环己二酮的含量为97.2%,收率为85.2%。
[0067]
对比例1
[0068]
(1)将浓度为5wt%的盐酸400g(0.548mol),降温至5℃;
[0069]
(2)利用上述步骤(1)的盐酸滴加加氢还原产物200g(加氢还原产物 中,1,3-环己二酮的含量30wt%,0.536mol),滴加至ph为2时,停止滴加, 加入1g晶种,控制温度保持在5℃下,搅拌2小时后,继续保温1小时,将 酸化结晶产物过滤、烘干,得到产品,其中,1,3-环己二酮的含量为92.1%, 收率为90.6%。
[0070]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在 本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包 括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样 应当视为本发明所公开的内容,均属于本发明的保护范围。
完整全部详细技术资料下载
当前第1页 1 2
相关技术
- 一类光诱导的细胞共价标记荧光...
- 一种无毒提取紫杉醇的生产装置...
- 香辛料油脂浸提池的制作方法
- 一种1,5-戊二胺的提取方法...
- 一种胆甾酸及其衍生物的除杂方...
- 一种环保型发胶及其制备方法和...
- 一种外泌体纯化方法及其一体机...
- 外泌体分离纯化检测方法及装置...
- 一种外泌体纯化方法及其装置与...
- 一种提高有机硅乳液稳定性的方...
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1