一种端粒酶启动基因表达方法及其应用与流程
本发明涉及属于医学基因治疗技术领域,具体涉及一种端粒酶启动基因表达方法及其应用。
背景技术:
癌症治疗技术现状:人类对于癌症治疗有最迫切的期望,科学界和医学界为此坚持不懈地付出探索。癌症是目前困扰人类健康威胁人类生命的重大疾病。虽经发病机理的大量基础研究和各种治疗方法的广泛临床实践,但截至目前,科学界和医学界还没有找到一种可治愈癌症的技术。虽然传统的手术切除加放化疗技术已被普遍用癌症治疗,以及最新的肿瘤免疫疗法也已被探索用于癌症治疗,但治疗的效果还与病人对于健康和生命的期望相差甚远。例如,目前应用最广泛且最有效的肿瘤免疫疗法是pd-1/pd-l1抑制剂(如pd-1抗体),但其目前的总体应答率却只有20-30%;而在血液恶性肿瘤治疗上有效的car-t、tcr-t等细胞治疗,在实体瘤治疗方面还没有实质进展。因此,探索新的癌症治疗技术,在人类可真正治愈癌症前,将始终是科学界和医学界努力的方向。
肿瘤治疗最关键和重要的找到在癌细胞表面表达的特定抗原。目前免疫治疗中不管是抗体治疗还是细胞治疗,都无法避免的依赖于该类肿瘤抗原(如目前car-t治疗中运用的最成功的cd16),即肿瘤免疫治疗的靶点。但目前发现的可资利用的这类抗原极其有限,且大多数在正常细胞上也低量表达,在应用时往往引起car-t细胞对正常细胞/器官的攻击,导致自身免疫症状。因此,目前该领域正在着力运用二代测序技术寻找更多的新抗原。此外,即使找到这类新抗原,car-t治疗的个体化细胞生产也因高昂的成本和潜在的新癌化风险严重阻碍其应用,因此不得不诉诸通用型t细胞的创制。但通用型t细胞也并非完全通用,而仅仅是一种可用于罹患某种同一类型抗原型肿瘤患者的t细胞,对于不同抗原型的肿瘤,仍需制备其特异的t细胞。尽管如此,该类通用型car-t的研制,仍代表了肿瘤免疫疗法最新的实用化探索。因此,医学界正努力实现通用型t细胞的研制和临床应用,并已取得显著进展。例如,最近(2017年3月)美国fda已经批准针对肿瘤抗原cd123的通用型car-tucart123(cellectis)进入临床试验。cd123抗原在急性骨髓性白血病(aml)细胞与母细胞性浆细胞样树突状细胞瘤(bpdcn)细胞上高度表达。这两种疾病都往往在骨髓中发病,且能在短期内威胁到患者的生命。通用型car-tucart123可用于治疗这两种癌症的免疫治疗。
必须指出,任何将病人细胞分离出来,进行体外培养、遗传操作等之后,再回输到病人体内的治疗方案,都存在体外操作带来的风险,尤其是遗传操作。
因此,癌症治疗技术的探索需要另辟蹊径的原创性设想。
基因编辑技术用于癌症治疗:近几十年来,随着全基因组测序技术的不断成熟,科学家们在各种细菌和古细菌(archaea)中陆续发现了很多成簇的、规律间隔的短回文重复序列(crispr序列)以及和crispr序列相关基因(cas)。研究发现,这些crispr序列与很多病毒或者质粒的dna序列互补,很多crispr-cas系统需要多种蛋白的参与,但只要一种细菌编码的内切酶cas9可以利用向导rna(核糖核酸)分子对特定的dna片段进行定向切割,这种基因组编辑技术就被称为crispr-cas9。crispr-cas9能够对基因组进行特异性定点改造,对细胞乃至整个生物体进行基因组改造。目前科学家已经利用这种技术对细菌、植物(农作物)、人体细胞以及斑马鱼进行过成功的遗传学改造工作。基因编辑无疑将成为下一代生物技术的核心。
基因编辑技术在人类遗传病及癌症治疗上具有巨大的应用价值。因此,各国科学家及医学界已着手其医疗价值的开发研究,如目前已经报道的用于治疗人镰刀型型贫血病、地中海型贫血、黄斑性失明等。目前已经报道,中美科学家开始将基因编辑技术用于癌症治疗。2016年,英国《自然》(nature)报道中国科学家已经开始将crispr-cas9基因编辑技术用于肿瘤car-t疗法(crisprgene-editingtestedinapersonforthefirsttime.naturedoi:10.1038/nature.2016.20988)。这是全球首例基因编辑技术治疗癌症的试验。根据《自然》杂志的介绍,卢铀团队将会从招募参加临床试验的患者血液中采集t细胞(一种免疫细胞),然后使用crisprcas9技术来敲除细胞中的一个基因,即pd-1蛋白编码基因。pd-1蛋白通常是细胞免疫反应的一个检查点,可以防止免疫细胞攻击健康的细胞。经过基因编辑的细胞会在实验室进行培养增殖,然后回输到患者的血液中。改造后的细胞将在患者体内巡视。卢铀表示,研究团队希望它们能对癌细胞发起攻击。目前,美国利用基因编辑技术进行癌症治疗的研究也已经得到批准进入实施。美国计划中的临床试验也打算敲除编码pd-1蛋白的基因,不过在将细胞回输给患者前,还会敲除另一个基因,再加入一个新的基因。虽然2015年美国fda已批准了两种基于抗体的阻断pd-1的疗法,用于治疗肺癌,但这些抗体会在多大程度上阻断pd-1并激活免疫反应,对每个患者来说都是难以预测的。与之相比,以敲除基因的方法阻断pd-1,其疗效应可能远高于抗体治疗。
但众所周知,crispr基因编辑技术有可能会在错误的位置编辑基因组,从而可能出现有害影响。虽然在将细胞回输给患者前可确认敲除的是正确的基因,但难以确保其他部分可能发生的所有错误编辑,这对该技术的应用埋下了隐患。此外,如上所述,任何将病人细胞分离出来,进行体外培养、遗传操作等之后,再回输到病人体内的治疗方案,都存在体外操作带来的风险。运用基因编辑技术操作病人离体细胞之后再回输,同样无法避免这种风险,甚至加重了这种风险。
因此,利用基因编辑技术探索癌症治疗新技术,不一定要死盯住基因编辑技术的最诱人却最具风险的编辑功能,可以充分开发利用其基因组dna靶向功能和功能改性价值,以此扬起优点避其弱点,探索利用基因编辑系统治疗癌症的新方法。
基因编辑工具的改性与基因表达调控:大量研究表明,并不是所有的疾病都是由于基因突变导致基因产物功能异常引起的,而是很多疾病是由于基因表达水平异常造成的。因此,调控基因的表达水平可用于治疗疾病。调控基因的表达水平有两种方法,一是向细胞导入目的基因的表达载体,使其在细胞表达产生基因产物(如rna或蛋白质),达到调高细胞内目标基因产物的目的。二是向细胞内导入具有基因表达调节功能的分子,使其激活或抑制细胞内源性目的基因的表达,达到调控细胞内目标基因产物的目的。后者调控目的基因关键要做到两点:一是靶向,即只调控目的基因,避免调控其他非目标基因;二是可激活或抑制基因表达。
目前,所有用于基因编辑的工具经过功能改性,均可以用于基因表达调控。例如,锌指蛋白(zincfingerprotein,zfp)、转录因子样效应物(transcriptionactivator-likeeffector,tale)、crispr等。在用于基因编辑时,前两者融合核酸酶内切酶(常用foki),以便切割dna,产生双链断裂,而crispr的cas9蛋白则自身具有核酸内切酶活性。在用于基因表达调控的激活时,前两者直接融合转录激活结构域(transcriptionalactivatingdomain,tad)(如vp16、vp64、vp48、vp160、p65、vpr等)或其他具有基因转录激活作用的结构(如p300、tet1等),而crispr的cas9蛋白则须突变掉内切酶活性(d10a&h840a)成为无核酸酶活性的cas9蛋白(nuclease-deficientcas9;也称为deadcas9,dcas9),并融合tad。在用于基因表达调控的抑制时,zfp、tale、dcas9均融合基因转录抑制结构域(transcriptionalinhibitingdomain,tid)(如kreb)或其他具有基因转录抑制作用的结构(如lsd1、dnmt3a等)。此外,zfp、tale、dcas9本身因结合dna而形成转录机器的路障,也可发挥基因表达调控的抑制作用(如crispri)。因此,当这些改性的基因编辑工具用于基因表达调控时,则成为人工转录因子(或称为工程化转录因子)。
除了对dcas9的上述改性外,还发展了dcas9-suntag的融合蛋白,该蛋白可与scfv-sfgfp-vp64融合蛋白结合,从而间接将转录激活蛋白vp64募集到靶基因上,激活靶基因的表达。该系统中,suntag是融合到dcas9上的肽表位(peptideepitope),而scfv-sfgfp-vp64是融合sfgfp(superfolder-gfp)和vp64的单链可变片段(single-chainvariablefragment,scfv)抗体。scfv抗体与肽表位可互作结合。通过融合多个串联肽表位,该系统可募集多达24个拷贝的scfv-sfgfp-vp64分子,可强烈激活内源基因表达。
此外,dcas9/sgrna用于基因表达调控时,除了上述对dcas9进行改性(如融合vp16、vp64、p65、vpr、p300、kreb等)外,还可以对sgrna进行改性,使其发挥基因表达调控作用。例如,在正常sgrna的3′末端添加发卡核酸配体(hairpinaptamer)序列[如tetraloop(sgrna1.1)、stemloop2(sgrna1.2)],通过ms2蛋白与发卡核酸配体的结合,将ms2-vp64或ms2-p65-hsf1等具有转录激活或抑制活性的融合蛋白募集到靶基因上,调控靶基因的表达(该系统被称为sam)。除了sam方式,还发展了多任务crisprrna脚手架(multi-taskingcrisprrnascaffolds)系统,即将模块化的脚手架rna(modularscaffoldrnas)(如ms2rna、pp7rna、comrna等)连接到正常sgrna上,通过脚手架rna募集具有不同功能的rna结合蛋白(即effectorprotein,效应物蛋白)(如ms2-mcp-vp64、pp7-pcp-vp64、com-com-vp64等),发挥基因表达调节的作用。
这些人工转录因子提供了人为干预基因表达的重要工具,在研究基因功能(基因过表达或敲低)、改造细胞功能(如细胞重编程)、疾病治疗(如肿瘤分化疗法)等领域具有重要应用价值。但目前还没有将这些人工转录因子成功用于基因治疗的实例。
端粒酶与癌症治疗:1985年,blackburn等从四膜虫首次分离出端粒酶。1989年,morin自真核细胞hela细胞分离出人端粒酶,发现其中用于合成端粒dna的rna模板为11nt的序列5′-ccuaacccuaac-3′,可指导合成大量的端粒dna重复序列5′-ttaggg-3′。
肿瘤研究中人们很早就发现端粒酶在绝大多数人类肿瘤细胞中被重新激活。被激活的端粒酶维持了肿瘤细胞的端粒长度,因此造成肿瘤细胞的无限增殖能力。在健康的机体中,端粒酶活性在绝大多数体细胞都中被编程性关闭,仅在胚胎组织、干细胞、生殖器官和一些急性再生组织(如肠上皮)中有活性,但在这种情况下,端粒酶活性通常远低于其在肿瘤细胞中的活性。大量的研究表明,医学界已经检测各种肿瘤组织的端粒酶活性,发现在各种恶性肿瘤组织中端粒酶检出率高达84%~95%,而正常组织和良性肿瘤中的检出率仅为1%~4%。因此端粒酶活性已经成为目前已知的最广谱和最典型的恶性肿瘤标志物。此外,研究发现在许多常见肿瘤的早期甚至是癌前阶段即可检测到端粒酶的活性。端粒酶活性的表达是癌变过程的一个早期事件,是许多恶性肿瘤发生的一个先决条件。近年来,端粒酶活性的检测已不仅仅局限于活检组织或切除标本,诸如分泌物、病理性体液、吸出物、冲洗物和血液等标本也得到了广泛的应用,并取得了满意的结果。例如,检测发现膀胱癌循环血肿瘤细胞的端粒酶活性100%、卵巢癌循环肿瘤细胞端粒酶活性100%、肝癌循环肿瘤细胞的端粒酶活性80%。因此,端粒酶活性不仅对肿瘤诊断特别是早期诊断具有重要意义,而且是优良的恶性肿瘤治疗靶点。正因如此,近年来如何抑制端粒酶活性不仅一直是肿瘤分子治疗学的研究热点,更有大量制药公司进行各种端粒酶活性抑制剂的开发,如反义基因、逆转录酶抑制剂、核苷类似物、核酶、蛋白激酶c抑制剂、诱导分化剂等。但截至目前,还没有一例端粒酶活性抑制剂成药。原因是各种端粒酶抑制剂均存在较严重的副作用,如对正常组织细胞产生的毒性、起效慢等。时至今日,端粒酶活性抑制在肿瘤治疗领域已近成鸡肋。
生物学研究表明,在机体内一种生物分子的活性很难在细胞特别是组织水平上被彻底扑灭。抑制从来不是解决问题的好办法,应如大禹治水,采用因势利导的策略,引导其发挥有利作用。因此,针对端粒酶活性这一最广谱和最典型的恶性肿瘤标志物,应创制新的肿瘤治疗策略。
端粒酶在80~90%的肿瘤类型存在激活,而正常的体细胞中没有端粒酶活性,因此端粒酶一直是公认的优良的肿瘤治疗靶点。据此,已有很多制药公司从抑制端粒酶活性抑制角度进行了大量的药物研发,但截至目前还没有一种端粒酶活性抑制剂成为可临床应用的药品。因此,针对端粒酶这一珍贵的恶性肿瘤遍在性靶点进行癌症的治疗,需要发展新的策略与技术。
技术实现要素:
发明目的:针对现有技术存在的问题,本发明名提供这一种端粒酶启动基因表达方法(telomerase-activatinggeneexpression,tage)。端粒酶可结合并延长dna分子末端的端粒酶识别序列,合成新的端粒重复序列;基于这一特性,将端粒酶特有的生物学功能与人工转录因子调控细胞基因表达技术相结合,发展了tage新方法。
本发明还提供了一种端粒酶启动基因表达方法的应用。
技术方案:为了实现上述目的,如本发明所述的一种端粒酶启动基因表达方法,包括如下步骤:
(1)将含有端粒识别序列的核酸分子与载体序列相连,构建出携带有端粒酶识别序列的效应基因表达载体并导入细胞,端粒酶在细胞内结合并延长该基因表达载体,在其末端合成的端粒重复序列;
(2)同时向细胞导入可识别结合端粒重复序列的基因表达激活系统;
(3)基因表达激活系统识别并结合端粒重复序列,激活效应基因表达载体上效应基因的表达。
其中,步骤(1)所述核酸分子带有位于其末端的粘性末端序列或带有由核酸酶切割后产生的粘性末端序列。核酸酶为直接转入细胞中的酵母同宗接合切换内切酶(homothallicswitchingendonuclease,ho酶)或转入细胞中的酵母同宗接合切换内切酶表达载体表达的内切酶(即转入细胞中的ho酶表达载体表达的ho酶)。
所述粘性末端序列含有一段3′突出的单链序列。
所述3′突出的单链序列含有端粒酶识别序列,如5′-tgtt-3′、5′-agtt-3′,或其他可被端粒酶识别的序列。
其中,步骤(1)所述载体序列含有一种或多种基因序列及基因表达所需元件;所述基因为其表达产物对细胞生理可产生影响,或致细胞死亡的基因;所述基因表达所需元件包括各种来源的最小启动子序列、kozack序列或polya序列。
所述基因包括cas9、tn5(转座酶5)、核酸内切酶、颗粒酶、凋亡酶(caspase)等基因。
其中,步骤(2)所述基因表达激活系统为基因转录激活分子或复合物,包括锌指蛋白(zfp)、转录激活物样效应物(tale)、crispr/cas9、端粒dna结合蛋白(telomere-bindingproteins,tbp)等;这些分子或复合物可识别结合端粒重复序列,并能激活效应基因表达载体上效应基因的表达。
所述crispr/cas9包括可靶向结合端粒重复序列及其附近序列的引导rna(guidedrna,grna)和无核酸酶活性的cas9(nuclease-deficientcas9;也称为deadcas9,dcas9),无核酸酶活性的cas9为dcas9。其中,所述引导rna包括单分子引导rna(single-guidedrna,sgrna)和双分子引导rna;其中,所述双分子grna由反式激活crisprrna(trans-activatingcrisprrna)和crisprrna(crisprrna,crrna)两种组分构成;其中,所述sgrna是tracrrna和crrna的人工嵌合体(也称为singlechimericguiderna);crispr/cas9系统指dcas9与sgrna的复合物,即sgrna-dcas9或sgrna/dcas9。
所述dcas9为融合蛋白,包括dcas9部分和融合蛋白部分。其中,dcas9部分包括目前已发现的经人工突变失去核酸内切酶活性的cas9蛋白,也包括其他具有类似功能的蛋白(如cpf1、c2c1、c2c2、c2c3、casx、casy、arman-1cas9、arman-4cas9等),以及其他新发现的类似功能物。
所述锌指蛋白、转录激活物样效应物蛋白和端粒dna结合蛋白为融合蛋白,包括锌指蛋白、转录激活物样效应物蛋白和端粒dna结合蛋白部分及融合蛋白部分。
所述融合蛋白部分包括各种目前已知转录激活结构域(transcriptionactivatingdomain,tad)(如p65、vp16、vp64、vpr等)、具有基因转录激活功能的其他蛋白(如p300、tet1等),以及其他新发现的类似功能物。
具有基因转录调节功能的分子或复合物,包括suntag系统、sam系统、脚手架rna系统等。suntag系统由dcas9-肽表位(peptideepitope)融合蛋白与单链可变片段抗体-转录激活结构域融合蛋白构成,例如dcas9-肽表位蛋白与scfv-sfgfp-vp64蛋白;其中scfv-sfgfp-vp64为融合sfgfp(superfolder-gfp)和vp64的单链可变片段(single-chainvariablefragment,scfv)抗体。其中肽表位也可与tale蛋白形成融合蛋白,实现suntag系统功能。sam系统和脚手架rna系统都由改造的引导rna和与改造的引导rna结合的蛋白构成。其中,改造的引导rna指通过对常规引导rna序列的加长,使引导rna分子上增加新的序列,这些序列可被其他具有转录激活功能的蛋白分子结合,如发卡核酸配体(hairpinaptamer)序列、脚手架rna(scaffoldrna,scrna)等。其中,与改造的引导rna结合的蛋白指一类可与改造的引导rna结合并且具有转录激活功能的融合蛋白分子,如ms2-vp64、ms2-p65-hsf1、ms2-mcp-vp64、pp7-pcp-vp64、com-com-vp64等。例如由发卡核酸配体tetraloop(sgrna1.1)、stemloop2(sgrna1.2)与其结合蛋白ms2-vp64、ms2-p65-hsf1等构成的sam系统。
基因转录激活分子或复合物,既可以是直接导入细胞的基因转录激活分子或复合物,如tale-vp64蛋白或sgrna/dcas9-vp64复合物,或基因转录激活分子或复合物的基因表达载体(包括dna或rna表达载体),例如向细胞导入tale-vp64或sgrna/dcas9-vp64的基因表达载体,这些载体在细胞内可表达产生tale-vp64蛋白或sgrna/dcas9-vp64复合物。
其中,步骤(3)所述效应基因的表达,其表达产物为rna或蛋白质。
所述rna为可引起细胞生理发生改变的rna。rna包括各种功能类型的rna(如microrna等),这些rna可引起细胞生理发生改变。
所述蛋白质为可引起细胞生理发生改变的蛋白质或多肽。蛋白质包括各种功能类型的蛋白质(如cas9、核酸内切酶、转座酶、颗粒酶、凋亡酶等)及多肽等;这些蛋白质及多肽可引起细胞生理发生改变,或致细胞死亡。
所述cas9蛋白与引导rna结合,靶向结合并切割细胞的端粒dna序列或其他dna序列,引发细胞生长抑制或死亡;所述tn5蛋白与转入细胞的含有me序列的dna结合,非靶向性切割细胞dna,引发生长抑制或细胞死亡;所述核酸内切酶特异性或非特异性切割细胞dna,引发细胞生长抑制或死亡;所述颗粒酶可引发细胞凋亡,如颗粒酶b;所述凋亡酶可引发细胞凋亡,如caspase9等。
本发明所述的端粒酶启动基因表达方法在制备杀灭肿瘤细胞的药物中的应用。
所述杀灭肿瘤细胞的药物为将端粒酶启动基因表达方法所需要的效应基因表达载体和可识别结合端粒重复序列的基因表达激活系统与病毒载体或纳米材料载体包装,制备成基因治疗药物;或将效应基因表达载体、可识别结合端粒重复序列的基因表达激活系统和ho酶或其表达载体,与病毒载体或纳米材料载体包装,制备成基因治疗药物;用于杀灭肿瘤细胞。
所述的肿瘤细胞包括体外培养的肿瘤细胞系/株等,如hepg2、a549、ht-29、hela或skov3细胞等,以及人体内自然发生的肿瘤细胞。
本发明基于端粒酶的生物学功能提出了一种被命名为tage(端粒酶启动基因表达方法telomerase-activatinggeneexpression,tage)的新技术。端粒酶的生物学功能是保护细胞的端粒不因dna复制而缩短,即端粒酶提供一种rna模板,使端粒末端突出的3′单链得以聚合延长,产生多重端粒重复序列(5′-ttaggg-3′)。本发明正是基于端粒酶的这一特性,将一段携带端粒酶识别3′突出单链的基因表达载体导入细胞,使端粒酶在细胞内结合并延长该基因载体,在其末端合成双链端粒重复序列;同时向细胞内导入一种基因表达激活系统(如sgrna-dcas9),使该基因表达激活系统识别并结合基因表达载体上被端粒酶合成的端粒重复序列,以此激活基因表达载体上的效应基因(如cas9)的表达。该效应基因的表达产物,将会对细胞的生理、生存产生严重影响,导致细胞生长抑制或死亡。例如,效应基因产物cas9蛋白在靶向端粒dna的sgrna的帮助下,切割细胞的端粒序列,造成端粒dna损伤,导致细胞死亡。
有益效果:本发明充分利用肿瘤细胞的端粒酶活性特征,将端粒酶特有的生物学功能(即端粒酶可结合并延长dna分子末端的端粒酶识别序列,合成新的端粒重复序列)与人工转录因子调控细胞基因表达技术相结合,发展了tage新技术。tage技术在细胞中可以有效发生,人工转录因子可激活效应基因在端粒酶阳性细胞中的表达,效应基因的表达产物可有效杀灭肿瘤细胞,本发明证明tage技术可用于杀灭肿瘤细胞,而对无端粒酶活性的细胞不产生影响。因此,tage技术应用在制备杀灭肿瘤细胞药物中,可用于癌症治疗。
附图说明
图1tage技术及其杀灭细胞的原理示意图;
图2转录激活物sgrna-dcas9用于tage技术及其杀灭细胞原理示意图;
图3酵母ho酶用于tage技术及其杀灭细胞原理示意图;
图4转录激活物tale用于tage技术及其杀灭细胞原理示意图;
图5细胞内端粒酶合成端粒dna实验;
图6tage技术激活效应基因gfp表达实验(293t细胞);
图7tage技术激活效应基因gfp表达实验(hepg2细胞);
图8tage技术激活效应基因gfp表达实验(mrc-5细胞);
图9tage技术杀灭肿瘤细胞实验(293t细胞);
图10tage技术杀灭肿瘤细胞实验(hepg2细胞);
图11tage技术杀灭肿瘤细胞实验(a549细胞);
图12tage技术杀灭肿瘤细胞实验(ht-29细胞);
图13tage技术杀灭肿瘤细胞实验(hela细胞);
图14tage技术杀灭肿瘤细胞实验(skov3细胞);
图15tage技术杀灭肿瘤细胞实验(mrc-5细胞);
图16tage技术杀灭肿瘤细胞实验(阿尔玛蓝分析);
图17靶向端粒dna的sgrna-cas9切割端粒实验(hepg2细胞);
图18靶向端粒dna的sgrna-cas9切割端粒实验(mrc-5细胞);
图19靶向端粒dna的sgrna-cas9切割端粒实验(293t细胞);
图20靶向端粒dna的sgrna-cas9切割端粒实验(阿尔玛蓝分析);
图21ho酶用于tage技术实验1(293t细胞);
图22ho酶用于tage技术实验2(293t细胞);
图23tale用于tage技术实验(293t细胞)。
具体实施方式
以下结合实施例和附图对本发明作进一步说明。
实施例1
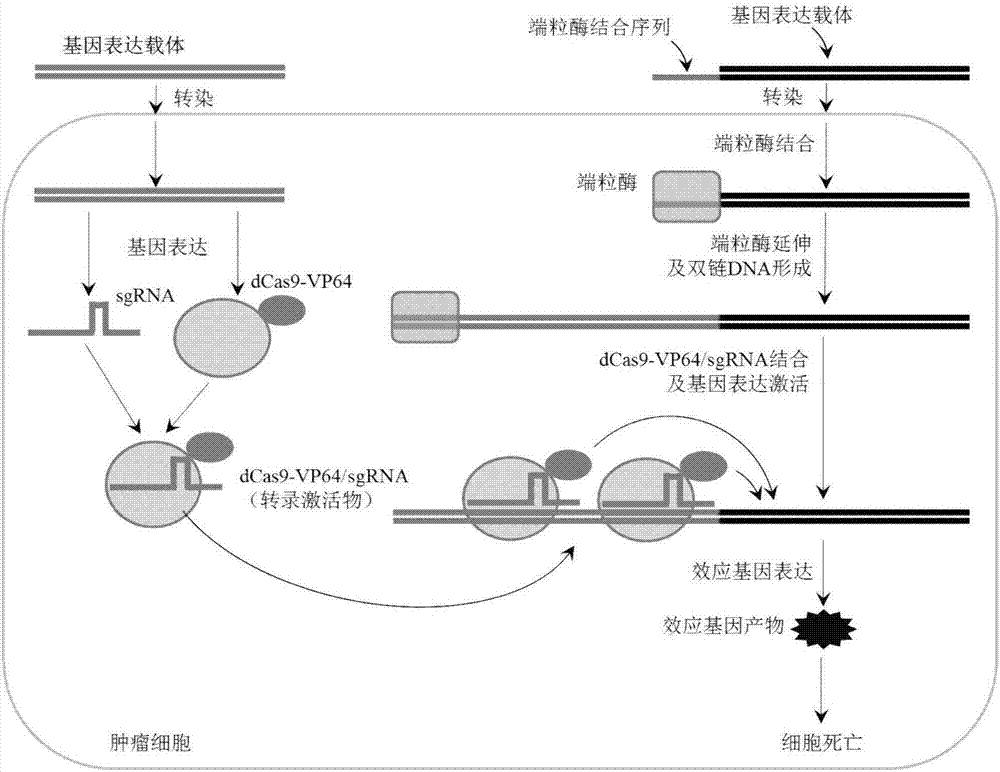
如图1所示,图示说明端粒酶启动基因表达方法的原理及流程。
端粒酶启动基因表达方法:
(1)将含有端粒识别序列的核酸分子与载体序列相连,构建出携带有端粒酶识别序列的基因表达载体并导入细胞,端粒酶在细胞内结合并延长该基因表达载体,在其末端合成的端粒重复序列;
(2)同时向细胞导入可识别结合端粒重复序列的基因表达激活系统;
(3)基因表达激活系统识别并结合端粒重复序列,激活基因表达载体上效应基因的表达。
实施例2
如图2所示,图示说明以crispr/cas9系统(sgrna/dcas9-vp64)为转录激活物实现端粒酶启动基因表达方法的原理及流程。
(1)将含有端粒识别序列的外源核酸分子与载体序列相连,构建出携带有酶端粒识别序列的基因表达载体并导入细胞,端粒酶在细胞内结合并延长该基因表达载体,在其末端合成的端粒重复序列;
(2)同时向细胞导入可识别结合端粒重复序列的基因表达激活系统sgrna/dcas9-vp64;
(3)sgrna/dcas9-vp64识别并结合端粒重复序列,激活基因表达载体上效应基因的表达。
实施例3
如图3所示,图示说明利用ho酶切割效应基因表达载体上的ho识别序列,以此实现端粒酶启动基因表达方法的原理及流程。
实施例4与实施例2步骤相同,不同在于步骤(2)导入细胞的基因表达载体除可表达转录激活物外,还可表达ho酶;所表达的ho酶可切割效应基因表达载体上的ho识别序列,产生端粒酶可识别的粘性末端。
实施例4
如图4所示,图示说明以tale系统(tale-vp64)为转录激活物实现端粒酶启动基因表达方法的原理及流程。
实施例3与实施例2步骤相同,不同在于步骤(2)导入细胞的基因表达激活系统为tale-vp64。
实施例5
1、细胞内端粒酶合成实验
本实验的目的是论证在人工导入细胞的dna分子的末端可否发生端粒酶合成。
构建载体:
[1]stmep:为一线性dna片段(线性表达载体),其序列构成为(5′→3′):端粒酶识别序列-最小启动子序列-egfp序列-pa(polya序列);其中端粒酶识别序列为3′突出单链,序列seqidno1为:3′-ttgagacgagctgcctaagg-5′。理论上端粒酶可结合到该载体的3′突出单链上,并由此聚合延长,合成由大量串联重复端粒dna构成的新的双链dna序列。
stmep:s,stickend(粘性末端);t,telomeraserecognitionsequence(端粒酶识别序列);m,minimalpromoter(最小启动子序列);e,egfp(绿色荧光蛋白编码序列);pa,polya(多聚a)。
[2]btmep:为一线性dna片段(线性表达载体),其序列同stmep,但3′端为平末端,无3′端端粒酶识别3′突出单链。该dna片段作为stmep的阴性对照。因无3′端端粒酶识别3′突出单链,端粒酶不能结合该载体,也无法合成新的端粒dna序列。所以该载体作为stmep的阴性对照。
btmep:b,bluntend(平末端)。
[3]sgrna:该实验中特指靶向人端粒dna序列的单引导rna(singleguidedrna,sgrna)。本实验中构建一单独真核表达载体(质粒),sgrna在细胞内由u6启动子负责转录合成。
[4]dcas9:该实验中指融合转录激活结构域vp64的突变掉内切酶活性的cas9蛋白。dcas9可视为一种可借助sgrna激活基因表达的人工转录因子。本实验中构建一单独真核表达载体(质粒),dcas9在细胞内由cmv(巨细胞病毒)启动子负责转录合成。
dcas9蛋白可与sgrna结合,形成sgrna-dcas9复合物,该复合物可借助sgrna识别并结合其靶序列(这里指人端粒dna序列)。
转染细胞:
本实验选择使用293t、hepg2、mrc-5三种细胞,选择依据为:
[1]hek293(humanembryonickidneycells293,人胚胎肾细胞)细胞比较容易转染,是一个很常用的表达研究外源基因的细胞株。293细胞系是原代人胚肾细胞转染5型腺病毒(ad5)dna的永生化细胞,表达转染的腺病毒5的基因。293t细胞表达e1a蛋白,s40大t抗原,含有s40复制起始点与启动子区的质粒可以复制。293t/17的转染效率更高,成为研究基因功能的一个强大工具。293t细胞是实验室中用于研究基因转染最常用的细胞,具有很高的转染效率;该细胞是人胚胎肾细胞转染大t抗原后形成的一种细胞,可用于腺病毒包装。因已有不少文献报道,293t细胞胞内具有端粒酶活性(据测定每个293t细胞约含有240个端粒酶单体;nucleicacidsresearch2014,42(13):8565–8577),本实验中视为具有端粒酶活性的非肿瘤细胞,同时用于监控每次的细胞转染实验试剂及操作。
[2]hepg2细胞是人肝癌细胞株(购于中科院上海细胞库),是肝癌研究中用得最广泛的细胞。经大量实验检测分析,其胞内具有很高的端粒酶活性,本实验中作为端粒酶阳性细胞。
[3]mrc-5是人胚肺成纤维细胞(购于中科院上海细胞库)。经大量实验检测分析,其胞内无端粒酶活性,本实验中作为端粒酶阴性细胞。
转染试剂:
脂质体(lipofectin),购于invitrogen。
实验操作:
[1]细胞培养:293t、hepg2、mrc-5细胞培养。细胞培养使用deme(hyclone;下同)(293t与hepg2)或rpmi1640(hyclone;下同)(mrc-5)培养基、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1小时(h)。分别用stmep、btmep、stmep+sgrna-dcas9转染三种细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。
[4]dna提取:胰酶消化收集各种处理的细胞,用基因组dna提取试剂盒(天根)按试剂盒说明提取各种处理的细胞的基因组dna(genomicdna,gdna)。
[5]dna质检:所提gdna的水平琼脂糖凝胶电泳检测及紫外分光光度(nanodrop)测定纯度、浓度。
[6]pcr扩增:用端粒dna检测引物(seqidno2:ts:5′-aatccgtcgagcagagtt-3′;seqidno3:cx:5'-cccttacccttacccttaccctaa-3′)、taq酶(takara)、taq酶缓冲液(takara)、dntp(各2.5mm,takara)、gdna建立pcr反应,并设置pcr阴性对照(不含gdna模板的pcr反应)。扩增程序:94℃5min;35个循环:94℃30s,58℃30s,72℃1min45s;72℃8min;4℃保存。
[7]dna电泳:将pcr产物用水平琼脂糖凝胶电泳,在对凝胶进行凝胶成像系统成像,获得电泳图片。
[8]dna测序:将水平琼脂糖凝胶上pcr扩增产物进行胶回收,做t载体克隆,用t载体通用引物进行序列测定。
实验结果:
实验结果如图5所示。
本实验采用的pcr引物为检测细胞端粒酶常用方法端粒重复序列扩增法(telomererepeatamplificationprotocol,trap)实验的pcr引物。trap实验中提供一dna片段,该片段的3′末端为人端粒酶可识别结合的单链区(seqidno4:5′-aatccgtcgagcagagtt-3′);检测细胞端粒酶活性时,首先将细胞的核蛋白提取出来,将trapdna片段与核蛋白共孵育,在端粒酶的作用下,trapdna片段末端合成新的端粒dna重复序列,之后用一对pcr引物(seqidno2:ts:5′-aatccgtcgagcagagtt-3′;seqidno3:cx:5'-cccttacccttacccttaccctaa-3′)对合成的dna进行pcr扩增,pcr产物用聚丙烯酰胺电泳检测,可看到梯状dna条带,据此判断细胞内的端粒酶活性。需说明的是,trap引物并不能扩增细胞本身的端粒dna,因为细胞本身的端粒dna上无trap引物之一(ts)的退火位点。同时证实若使用对照细胞(图5,lipofectin)的gdna进行trap引物的pcr扩增,不会产生pcr产物。
本实验将3′末端带有人端粒酶识别单链区的已线性dna,即stmep,转入细胞,若细胞内有端粒酶活性,且端粒酶成功识别stmepdna,并在该dna的末端合成新的端粒dna重复序列,则提取细胞dna后用trap引物进行扩增,则应扩增出端粒dna。若细胞内无端粒酶活性,或端粒酶未成功识别stmepdna并在其末端合成新的端粒dna重复序列,则扩增不出产物。
实验结果表明,凡用导入stmepdna(stmep、stmep+sgrna-dcas9)的端粒酶阳性细胞(293t与hepg2)的gdna做模板时,均扩增出产物(图5),产物在琼脂糖凝胶上呈弥散状(smear),这与常规trap检测结果是一致的。而作为stmep对照的btmep,引物端粒酶合成位点,既是转染端粒酶阳性细胞(293t与hepg2),也不产生pcr产物。这与实验预期是一致的。此外,当转染端粒酶阴性细胞(mrc-5)时,所有转染均不产生pcr产物。
pcr扩增产物的测序分析表明,pcr扩增产物确是人端粒dna重复序列,进一步说明在stmepdna的末端发生了胞内端粒酶合成反应。
这些结果,充分说明本实验设计的人工导入细胞的dna分子的末端可发生端粒酶合成。这种合成仅发生在端粒酶阳性细胞内,是一种端粒酶活性特异的胞内dna合成反应。
实施例6
端粒酶启动的基因表达系统验证
本实验的目的是论证tage技术能否在真核细胞内发生。
构建载体:
[1]stmep:同实施例5。
[2]btmep:同实施例5。
[3]sgrna:同实施例5。
[4]dcas9:同实施例5。
[5]c1-egfp:为一真核表达载体,在cmv启动子作用下表达绿色荧光蛋白。该载体在本实验中用于监控转染,以表明转染试剂和操作正确、成功。
转染细胞:
本实验仍使用293t、hepg2、mrc-5三种细胞,理由同实施例5。
转染试剂:
脂质体(lipofectin),购于invitrogen。
转染实验:
[1]细胞培养:293t、hepg2、mrc5细胞培养。细胞培养使用deme(293t、hepg2)或rpmi1640培养基(mrc5)、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、stmep、stmep+sgrna、stmep+dcas9、btmep+sgrna-dcas9、stmep+sgrna-dcas9转染三种细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
[4]流式测定:胰酶消化收集各种处理的细胞,用流式仪检测分析细胞的荧光有无及强度。
实验结果:
实验结果如图6~8所示。
上面的实验表明,只要人工导入细胞的dna分子的末端具有端粒酶识别序列,则细胞内端粒酶可结合该端粒酶识别序列,进而合成出新的端粒重复序列。在上面所用的stmep分子上,在端粒酶识别序列(seqidno1:3′-ttgagacgagctgcctaagg-5′)后,设计了一个最小启动子序列,其序列seqidno5为:5′-ttcgcatattaaggtgacgcgtgtggcctcgaacaccgagcgaccctgcagcgacccgcttaa-3′。最小启动子序列提供了tata盒等基本元件,为rna聚合酶ii(polii)与基础转录因子(如tfiid等)的结合部分。
在本实验中,设计了一个靶向人端粒dna序列的人工转录因子系统,即向人端粒dna序列的sgrna(靶序列seqidno6为:3′-atcccaatcccaatcccaat-5′)与dcas9;当将sgrna与dcas9表达载体导入细胞后,表达产生sgrna-dcas9复合物,该复合物可识别结合端粒dna序列,如stmep上新合成的端粒dna。当sgrna-dcas9复合物与stmep上新合成的端粒dna结合时,则dcas9蛋白上融合的转录激活结构域vp64可作用于最小启动子上结合的基本转录机器(polii与基础转录因子的复合体),激活最小启动子下游基因(本实验中编码绿色荧光蛋白egfp的基因)的表达,使共导入stmep、sgrna及dcas9分子的细胞产生绿色荧光。
而导入对照载体btmep的细胞,即使同时导入sgrna与dcas9表达载体(btmep、sgrna、dcas9共转染),也因无法合成新的端粒dna序列,btmep载体上不能出现sgrna-dcas9的靶点,而无法激活下游基因egfp的表达,细胞因此不产生绿色荧光。此外,理论上,只有stmep、sgrna及dcas9三种载体分子共同转染的细胞才会产生绿色荧光,而导入stmep与sgrna、stmep与dcas9的细胞都不会产生绿色荧光。
本实验结果表明,共转染stmep、sgrna及dcas9载体的细胞中,端粒酶阳性细胞hepg2与293t细胞中均出现绿色荧光,而端粒酶阴性细胞mrc-5不出现绿色荧光(图6~8)。在三种细胞中,其他对照转染(包括btmep+sgrna-dcas9,stmep+sgrna,stmep+dcas9)均不产生绿色荧光(图6~8)。实验结果符合预期,即tage技术可在端粒酶阳性细胞中表达目的基因,而在端粒酶阴性细胞中不表达目的基因。
实施例7
端粒酶启动的基因表达系统杀灭肿瘤细胞验证,tage技术及其杀灭细胞的原理示意图如图1所示。
本实验的目的是论证端粒酶活性特异的基因转录表达系统能否用于杀灭肿瘤细胞。
构建载体:
[1]stmcp:为一线性dna片段(线性表达载体),其序列构成为(5′→3′):端粒酶识别序列-最小启动子序列-cas9编码序列-pa(polya序列)。该线性载体相当于将stmep中的egfp编码序列替换为cas9蛋白编码学列,其他序列解结构完全同stmep载体。
stmcp:s,stickend(粘性末端);t,telomeraserecognitionsequence(端粒酶是识别序列);m,minimalpromoter(最小启动子序列);c,cas9(cas9蛋白编码基因);p,polya(多聚a)。
[2]btmcp:为一线性dna片段(线性表达载体),其序列结构及组成同stmcp,但3′端为平末端,无3′端端粒酶识别3′突出单链。该dna片段作为stmcp的阴性对照。因无3′端端粒酶识别3′突出单链,端粒酶不能结合该载体,也无法合成新的端粒dna序列。因此,sgrna-dcas9-vp64复合物在该载体上没有结合靶点,不结合该载体,从而无法启动下游报告基因的转录,则细胞不显示绿色荧光。所以该载体作为stmcp的阴性对照。btmcp:b,bluntend(平末端)。
[3]sgrna:同实施例5。
[4]dcas9:同实施例5。
[5]c1-egfp:同实施例6。
转染细胞:
本实验使用293t、hepg2、a549、ht-29、hela、skov3及mrc-57种细胞,选择依据为:
293t(肾上皮细胞)、hepg2(肝癌细胞)、mrc-5(胚肺成纤维细胞)的使用理由同上。hela为人宫颈癌细胞、skov3为人卵巢癌细胞、a549为非小细胞肺癌细胞、ht-29为人结肠癌细胞。这些癌细胞代表了常见的不同脏器的癌症。
转染试剂:
脂质体(lipofectin)。
实验操作:
[1]细胞培养:293t、hepg2、a549、ht-29、hela、skov3及mrc5细胞培养。细胞培养使用deme(293t、hela、hepg2)或rpmi1640培养基(a549、ht-29、skov3、mrc5)、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、stmcp、stmcp+sgrna、stmcp+dcas9、btmcp+sgrna-dcas9、stmcp+sgrna-dcas9转染上述细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
[4]细胞染色:吖啶橙染色按吖啶橙产品说明(索莱宝;北京)进行(室温染色10min)。染色后细胞用pbs洗涤。
[5]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
[6]增殖测定:按上述程序,重新培养、转染一批细胞,将细胞增殖检测试剂阿尔玛蓝(alamarblue)(购自上海翊圣生物科技有限公司;yeasen)在转染后4h加入细胞培养液(100μl培养基加10μl阿尔玛蓝),细胞转染后继续培养至转染后24h,用酶标仪(biotek)测定荧光强度(激发:530nm;发射:590nm);以阿尔玛蓝产品说明书计算各种处理细胞的增殖率。
实验结果:
实验结果如图9~16所示。
上面的实验表明,tage技术可在端粒酶阳性细胞中表达效应基因,而在端粒酶阴性细胞中不表达效应基因。外源目的基因的表达赋予端粒酶阳性细胞一种新的性状,例如在该实验中就是端粒酶阳性细胞产生绿色荧光。因此,随着外源效应基因的表达,端粒酶阳性细胞会产生新性状,如致死。恶性肿瘤细胞就是典型的端粒酶阳性细胞,让恶性肿瘤细胞生长抑制或致死,是医学治疗癌症中施以千方百计欲达成的目的。因此,期望用上述实验已经证明的tage技术抑制肿瘤细胞的生长或致其死亡。
在本实验中,上述stmep载体的基础上,将egfp基因替换为cas9蛋白基因,构建了stmcp载体。当将stmep、sgrna与dcas9载体分子共转染细胞后,sgrna与dcas9载体表达产生sgrna-dcas9复合物可与stmcp上新合成的端粒dna发生结合。dcas9蛋白上融合的转录激活结构域vp64继而作用于最小启动子上结合的基本转录机器(polii与基础转录因子的复合体),激活最小启动子下游cas9基因的表达,使共导入stmcp、sgrna及dcas9载体分子的细胞产生cas9蛋白。该蛋白可与细胞中已表达的靶向端粒dna的sgrna结合,形成靶向端粒dna的sgrna-cas9复合物。该复合物与细胞端粒dna解结合可导致端粒dna的切割,造成端粒dna损伤,从而引发细胞死亡。
而导入对照载体btmcp的细胞,即使同时导入sgrna与dcas9表达载体(btmcp、sgrna、dcas9共转染),也因无法合成新的端粒dna序列,btmcp载体上不能出现sgrna-dcas9的靶点,而无法激活下游基因cas9的表达,细胞因此不产生cas9蛋白,细胞相应也不会死亡。此外,理论上,只有stmcp、sgrna及dcas9三种载体分子共同转染的细胞才发生死亡,而导入stmcp与sgrna、stmcp与dcas9的细胞都不发生死亡。
本实验结果表明,共转染stmcp、sgrna及dcas9载体的细胞中,端粒酶阳性细胞hepg2、a549、ht-29、hela及skov3均发生显著死亡,而端粒酶阴性细胞mrc-5不发生死亡(图9~16)。在这些细胞中,其他对照转染(包括btmcp+sgrna-dcas9,stmcp+sgrna,stmcp+dcas9)均不发生死亡(图9~16)。用吖啶橙染色显微观测的结果与阿尔玛蓝(alamarblue)定量测定的结果一致。实验结果符合预期,即端粒酶活性特异的基因转录表达系统可用于杀灭肿瘤细胞。
本实验中,表现端粒酶阳性的293t细胞并未发生显著的死亡。因hek293细胞是来源于体外培养的正常人胚肾组织,后再传代培养中进行了大量的腺病毒转染,获得可连续传代的293细胞系,此期间的病毒转染可能激活了其端粒酶活性,但其端粒dna结构可能与恶性肿瘤细胞仍存在很大的差异,因此未被本实验所使用的系统所杀灭。这从另一个侧面反映,本实验所使用的系统具有很好的恶性肿瘤细胞特异性。此外,人体内也自然存在一些端粒酶活性阳性的非肿瘤细胞,如干细胞、分裂旺盛的上皮细胞等,293t细胞的表现,可能预示本实验所使用的肿瘤杀灭系统不会对这些非肿瘤细胞产生显著影响。
293细胞的缺陷是生长过程中贴壁强度比较小。所以在实验过程中容易流失,从而影响实验结果。实验中,发现当用吖啶橙色时,会导致293t大量脱壁,因此未获得293t细胞的吖啶橙色图像(图9)。
实施例8
利用crispr/cas9系统(即sgrna-dcas9)直接切割端粒dna实验,转录激活物sgrna-dcas9用于tage技术及其杀灭细胞原理示意图如图2所示。
本实验的目的是论证端粒酶活性特异的基因转录表达系统能否用否用于杀灭肿瘤细胞。
构建载体:
[1]sgrna-cas9:为一质粒表达载体,其中有u6启动子控制的靶向人端粒dna序列的sgrna表达序列,以及cmv启动子控制的cas9-2a-egfp蛋白表达序列。sgrna靶序列seqidno7为:5′-taaccctaaccctaacccta-3′;sgrna序列seqidno8为:5′-tagggttagggttagggtta-3′。
[2]c1-egfp:同实施例6。
转染细胞:
293t、hepg2、mrc5。选择原因同实施例5。
转染试剂:
脂质体(lipofectin)。
实验操作:
[1]细胞培养:293t、hepg2及mrc5细胞培养。细胞培养使用deme(293t、hepg2)或rpmi1640培养基(mrc5)、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、sgrna-cas9转染上述细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
[4]细胞染色:吖啶橙染色按吖啶橙产品说明(索莱宝;北京)进行(室温染色10min)。染色后细胞用pbs洗涤。
[5]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
实验结果:
实验结果如图17~20所示。
本实验中向细胞直接导入一端粒dna靶向sgrna和cas9蛋白共表达质粒载体,该载体可在细胞内表达sgrna及dcas9蛋白,二者形成靶向端粒dna的sgrna-cas9复合物。该复合物与细胞端粒dna结合可导致端粒dna的切割,造成端粒dna损伤,从而引发细胞死亡。
本实验结果表明,转染sgrna-cas9共表达载体的细胞中,端粒酶阳性细胞hepg2发生显著死亡,而端粒酶阴性细胞mrc-5不发生死亡(图17~18)。这种情况与上面的端粒酶活性特异基因表达系统取得的结果是一致,进一步说明上面的端粒酶活性特异基因表达系统作用的机理就是通过造成端粒dna的切割损伤而引发细胞死亡。
与上一个实验相似的是,转染sgrna-cas9共表达载体的端粒酶阳性293t细胞也未发生显著的死亡(图19)。这一结果证明了,即正常肾上皮细胞的293t细胞,即使其端粒酶活性因病毒感染等激活,但其端粒dna结构与恶性肿瘤细胞存在很大的差异,因此不能被靶向端粒dna的sgrna-cas9复合物有效切割而致细胞死亡。本实验进一步说明端粒酶活性特异基因表达系统所表达产生的端粒dna靶向sgrna-cas9复合物,对肿瘤细胞具有特异性,而对具有端粒酶活性的非肿瘤正常人体细胞不产生显著影响。
在三种细胞中,用吖啶橙染色显微观测的结果(图17~19)与阿尔玛蓝定量测定的结果(图20)是一致的。
实施例9
hosite-tmep转染实验,酵母ho酶用于tage技术及其杀灭细胞原理示意图如图3所示。
本实验的目的是论证ho酶产生的粘性末端用于tage技术。
构建载体:
[1]shosite-tmep:末端含有ho酶切点3′单链突出的(3′-ttgt-5′)的线性载体,其他结构及序列同上述stmep载体,如最小启动子序列、egfp序列、polya序列。
[2]bhosite-tmep:含有平末端(将shosite-tmep的3′单链突出序列3′-ttgt-5′转换为双链)、最小启动子序列、egfp序列及polya序列的线性载体。本实验中作为shosite-tmep载体的阴性对照。
[3]sgrna:同实施例5。
[4]dcas9:同实施例5。
转染细胞:
293t细胞。
转染试剂:
脂质体(lipofectin)
实验操作:
[1]细胞培养:293t细胞培养。细胞培养使用deme培养基、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、shosite-tmep、shosite-tmep+sgrna、shosite-tmep+dcas9、bhosite-tmep+sgrna-dcas9、shosite-tmep+sgrna-dcas9转染293t细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
实验结果:
实验结果如图21所示。
酵母同宗接合切换内切酶(homothallicswitchingendonuclease,ho酶)切割其dna靶点序列(如下)可产生3′突出单链区,且该3′突出单链区的3′末端含有3′-ttg-5′,这与上面的实验中所使用的trap引物(ts)末端的3′-ttg-5′相同。因此,本实验的目的是论证ho酶产生的粘性末端可否作为端粒酶识别序列用于tage技术。
ho内切酶的识别序列及其切割后产生的粘性末端如下:
seqidno9:5′-tttatgggactacttcgcgcaaca↓gtataa-3′
seqidno10:3′-aaataccctgatgaagcgcg↑ttgtcatatt-5′(↑,↓:断裂点)
结果表明,ho酶产生的粘性末端可作为端粒酶识别序列,用于tage技术。
本实验的目的是为tage技术的体内应用做出探讨。病毒载体(如腺病毒、慢病毒、腺相关病毒)常用体内基因转染,是体内应用中常用的基因治疗载体。但在体内使用中,由于粘性末端的线性载体(如上述stmep、stmcp)难以包装进病毒载体,这种情况下,可将ho内切酶表达载体、含有ho内切酶的识别序列的效应基因(如上面已探讨的cas9)表达载体、sgrna-dcas9表达载体等,包装进病毒载体,以便用病毒载体将tage技术所用分子导入体内细胞,使其在人体内细胞中发挥作用。
当然,在纳米载体业已成熟并已大行其道的今天及将来,运用纳米载体将使用粘性末端线性载体的tage技术所用分子导入体内细胞并不困难。本实验所进行的ho酶实验,提供了一种不使用纳米载体,而使用传统基因治疗载体——病毒载体,以实现tage技术体内应用的手段。
实施例10
hosite-tmep转染实验,酵母ho酶用于tage技术及其杀灭细胞原理示意图如图3所示。
本实验的目的是论证ho酶表达载体在细胞内可表达ho酶并可以产生粘性末端,该粘性末端可用于实现tage技术。
构建载体:
[1]shosite-tmep:同实施例9。
[2]bhosite-tmep:末端含有ho酶切点的平末端线性载体,其他结构及序列同上述stmep载体,如最小启动子序列、egfp序列、polya序列。最小启动子序列前为双链平末端ho内切酶识别序列。ho内切酶的识别序列及其切割后产生的粘性末端见实施例9实验结果部分的阐述。
[3]sgrna:同实施例5。
[4]dcas9:同实施例5。
[5]c1-ho:cmv启动子控制的ho酶表达载体。
转染细胞:
293t细胞。
转染试剂:
脂质体(lipofectin)
实验操作:
[1]细胞培养:293t细胞培养。细胞培养使用deme培养基、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、shosite-tmep、shosite-tmep+sgrna、shosite-tmep+dcas9、shosite-tmep+sgrna-dcas9、bhosite-tmep+sgrna-dcas9、bhosite-tmep+sgrna-dcas9+c1-ho、c1-ho转染293t细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
实验结果:
实验结果如图22所示。
结果表明,当细胞内转染ho酶表达载体时,可在细胞内产生ho酶,并且该酶切割bhosite-tmep载体,产生了shosite-tmep载体。shosite-tmep载体再在sgrna-dcas9的作用下,激活效应基因egfp的表达。可见,ho酶可切合产生粘性末端,该末端可作为端粒酶识别序列,用于tage技术。
本实验的目的同实施例9,是为tage技术的体内应用做出探讨。
实施例11
人工转录因子tale在tage技术中的应用,转录激活物tale用于tage技术及其杀灭细胞原理示意图如图4所示。
本实验的目的是论证人工转录因子tale可否用于tage技术。
构建载体:
[1]stmep:同实施例5。
[2]tale:可靶向结合人端粒dna序列的tale表达载体(质粒)。其人端粒dna结合靶点为:seqidno11:5′-tagggttagggttagggt-3′。
[3]c1-egfp:同实施例6。
转染细胞:
293t细胞。
转染试剂:
脂质体(lipofectin)
实验操作:
[1]细胞培养:293t细胞培养。细胞培养使用deme培养基、胎牛血清(hyclone)、5%co2等标准细胞培养条件;细胞复苏后,按同等密度(0.5×105/孔)接种到24孔微孔板中,培养过夜贴壁后,进行转染。
[2]细胞转染:细胞培养液换为无血清培养基培养1h。分别用c1-egfp、tale、stmep、tale+stmep转染293t细胞。空脂质体转染的细胞作为转染对照。每孔细胞的总dna用量及脂质体用量参考脂质体产品说明书进行。dna-脂质体加入无血清培养基培养4h。换为含血清新鲜培养基,继续培养24h。
[3]细胞观察:用倒置荧光显微镜(olympusix51-dpi71)观测拍照各种处理的细胞,主要观察细胞是否产生绿色荧光;同时观察细胞生长情况,如生长旺盛、贴壁良好、无污染等。对各种处理的细胞进行多视野明场及绿色荧光蛋白观察通道的照相拍摄。
实验结果:
实验结果如图23所示。
融合了转录激活结构域(如vp64、p65等)的锌指酶(znf)、转录因子样效应物(tale)及crispr/cas9(sgrna-dcas9)是近年来广泛使用的人工转录因子,被用于激活细胞内内源性基因的表达。目前,由于crispr(sgrna-dcas9)的简便性,在该领域使用最流行。上面的实验已经证实,sgrna-dcas9人工转录因子可用于tage技术。tale是继znf之后crispr产生之前而广泛使用的转录激活物,由于在某些方面仍较crispr有优势,目前该技术仍然尚有应用。因此,本实验的目的是论证人工转录因子tale可否用于tage技术。
结果表明,人工转录因子tale可否用于tage技术(图23)。靶向于端粒dna序列的tale蛋白成功激活了报告基因egfp的表达(图23)。
sequencelisting
<110>东南大学
<120>一种端粒酶启动基因表达方法及其应用
<130>2017
<160>11
<170>patentinversion3.3
<210>1
<211>20
<212>dna
<213>人工合成
<400>1
ttgagacgagctgcctaagg20
<210>2
<211>18
<212>dna
<213>人工合成
<400>2
aatccgtcgagcagagtt18
<210>3
<211>24
<212>dna
<213>人工合成
<400>3
cccttacccttacccttaccctaa24
<210>4
<211>18
<212>dna
<213>人工合成
<400>4
aatccgtcgagcagagtt18
<210>5
<211>63
<212>dna
<213>人工合成
<400>5
ttcgcatattaaggtgacgcgtgtggcctcgaacaccgagcgaccctgcagcgacccgct60
taa63
<210>6
<211>20
<212>dna
<213>人工合成
<400>6
atcccaatcccaatcccaat20
<210>7
<211>20
<212>dna
<213>人工合成
<400>7
taaccctaaccctaacccta20
<210>8
<211>20
<212>dna
<213>人工合成
<400>8
tagggttagggttagggtta20
<210>9
<211>30
<212>dna
<213>人工合成
<400>9
tttatgggactacttcgcgcaacagtataa30
<210>10
<211>30
<212>dna
<213>人工合成
<400>10
aaataccctgatgaagcgcgttgtcatatt30
<210>11
<211>18
<212>dna
<213>人工合成
<400>11
tagggttagggttagggt18
- 一种菊粉酶基因及其应用的制造...
- 甘草酸生物合成相关的糖基转移...
- 参与甘草酸生物合成的UGT基...
- 参与甘草酸生物合成的糖基转移...
- 氮营养运输基因OsNPF8....
- 一种烟草KC1基因及其制备方...
- 一种烟草AKT1基因及其制备...
- 重组人成纤维细胞生长因子‑1...
- 调控基于TOLL‑样受体的免...
- 一种水稻诱导抗病基因OsAA...
- 还没有人留言评论。精彩留言会获得点赞!
- 茶树nrt1基因、蛋白及基因表达方法
- CL5547.Contig2基因在南瓜基因表达实时荧光定量PCR分析中作为内参基因的用图
- 用于增强植物中组成型基因表达的调节性核酸分子的制作方法
- 检测eps8l3基因表达的试剂在制备骨质疏松症诊断产品中的应用
- 检测eapp基因表达的试剂在诊治阿尔茨海默病中的应用
- 抑制Grb2基因表达的siRNA及其应用
- 监督基因表达数据分类方法
- 一种pcr法检测人ercc1基因表达水平的试剂盒的制作方法
- 一种检测尿液中pca3基因和psa基因表达量的荧光定量pcr法及其诊断试剂盒的制作方法
- 基因表达的定量的制作方法
- 基于lspr检测精子顶体酶活性的方法、试剂盒及其应用
- 具有成骨细胞增殖促进活性的肽及其利用
- 人类羊膜间充质细胞体外诱导分化为胰岛素分泌细胞方法
- 用于治疗和预防良性前列腺增生的组合物的制作方法
- 用于药物活性试剂的原位释放的滞留装置和系统的制作方法
- 产生端粒酶逆转录酶(tert)的新方法
- Sirt6的调节物及其筛选分析的制作方法
- 检测tert基因启动子c250t、c228t突变位点的方法、引物和试剂盒的制作方法
- 神经胶质瘤和肿瘤亚群中tert启动子突变的制作方法
- Sirt6的调节物及其筛选分析的制作方法
- 用于端粒酶抑制的改性寡核苷酸的制作方法
- 端粒酶抑制剂及其制备方法和用途的制作方法
- 端粒酶活化化合物及其使用方法
- 端粒酶活化化合物及其使用方法
- 一种调控端粒酶活性与端粒长度、延缓细胞衰老、延年益寿的制剂及制备方法
- 用于端粒酶抑制的改性寡核苷酸的制作方法
- 端粒酶抑制剂及其使用方法
- 控制端粒长度的方法
- 端粒酶活化化合物及其使用方法
- 端粒酶阳性细胞中端粒长度的调节和癌症治疗的制作方法
- 端粒酶活化化合物及其使用方法
- 端粒酶活化化合物及其使用方法
- 一种调控端粒酶活性与端粒长度、延缓细胞衰老、延年益寿的制剂及制备方法
- 用于端粒酶抑制的改性寡核苷酸的制作方法
- 端粒酶抑制剂及其使用方法
- 端粒酶活化化合物及其使用方法
- 端粒酶阳性细胞中端粒长度的调节和癌症治疗的制作方法
- 抑制端粒酶活性脱氧核酶寡核苷酸结构及用途的制作方法
- 来自端粒酶的抗原性肽的制作方法
- 哺乳动物端粒酶的制作方法