一种基于深度学习的蛋白质药物结合位点预测方法与流程
本发明属于结合位点预测方法领域,特别涉及一种基于深度学习的蛋白质药物结合位点预测方法。
背景技术:
目前,随着新型疾病的出现以及病毒、细菌等病原体耐药性的提高,药物的研发面临着巨大的压力。药物的研发需要经历先导化合物的发现、临床试验、上市以及追踪等多个过程,需要耗费大量的人力、物力、财力以及时间。计算机辅助药物设计的出现一定程度上解决了部分问题,计算机辅助药物设计通过使用计算机模拟药物的代谢、筛选等过程,为药物研发提供帮助。在计算机辅助药物设计中,虚拟筛选是关键的步骤,其通过筛选能与靶点蛋白相结合的小分子化合物,为先导化合物的发现奠定基础。而结合位点的预测,是通过发现和预测靶点蛋白上与小分子结合的口袋,在虚拟筛选中具有重要的指导作用。
结合位点(bindingsites),也称为口袋(pockets),是蛋白质表面的空腔,是能与小分子化合物结合的作用位点。结合位点的定位,能为分子对接时的结合构象提供重要的指导作用,减少对接构象空间搜索区间,从而减少对接和虚拟筛选的时间。
结合位点的筛选,主要通过影响结合位点形成的几个关键因素来预测。蛋白质的几何结构是形成结合位点的一个重要因素,其表面的空腔往往是结合位点形成的绝佳区域。通过查找蛋白质的空腔或者是凹陷部位来寻找蛋白质的结合位点称为基于几何结构的方法(geometry-basedmethod)。ligsitecsc和ligsite通过将蛋白质离散为
非键相互作用(non-bondedinteractions)也是影响结合位点形成的重要因素,比如范德华力、氢键等,这类方法也成为基于能量的方法。基于能量的方法大多采用基团探针来不断计算各个位置与蛋白质的能量值,进而通过找出那些潜在的能量特异点来探索结合位点。q-sitefinder方法用-ch3探针来计算蛋白质网格的非键相互作用,并采用聚类算法对最终的能量分布进行聚类,预测出潜在的口袋。
蛋白质的残基序列中包含了丰富的遗传信息,而残基序列也是决定蛋白质功能的关键因素。通过挖掘蛋白质残基序列中所蕴含的信息来寻找结合位点称为基于序列的方法。该类方法只对蛋白质的残基序列进行分析,寻找具有结合活性的残基,其往往需要借助序列分析中的方法或者工具来协助分析。osml方法便是对蛋白质序列提取pssm(positionspecificscoringmatrix)并构建svm模型,其创新之处在于其构建模型的数据是根据输入的不同动态改变,也就是其所谓的query-drivendynamic。
随着机器学习尤其是深度学习在图形领域的广泛应用,其在药物设计领域也成为了研究的热点。比如deepsite,该方法根据原子类型构建蛋白质三维结构的8通道特征作为输入,进行卷积神经网络的训练,最终预测口袋的位置。
但是,基于几何结构的方法通过寻找蛋白质表面的空腔来寻找结合位点,也就是该方法只考虑蛋白质的几何结构。但在分子对接过程中,不少对接属于柔性对接,也就是在对接过程中,蛋白质与小分子接触时才产生口袋。因此,对于基于几何结构的方法来说,很难寻找适应于柔性对接的口袋,因而存在着局限性。
基于能量的方法则通过不断放置探针位置,来寻找其与蛋白质的能量值。该方法有效克服了基于几何结构中的弊端,能探索到适应于柔性对接的口袋。但其也忽视了蛋白质的立体结构,同样存在着局限性。
基于序列的方法对蛋白质的残基序列进行分析。蛋白质的序列蕴含了丰富的遗传信息,而序列同样也一定程度上决定了蛋白质的功能。但其忽视了蛋白质的几何结构和非键相互作用等影响蛋白质结合位点的重要因素。
技术实现要素:
本发明提出一种基于深度学习的蛋白质药物结合位点预测方法,能够综合考虑结合位点形成因素,基于深度学习进行结合位点的定位和预测。
本发明的技术方案是这样实现的:一种基于深度学习的蛋白质药物结合位点预测方法,包括如下步骤:
步骤1、选取蛋白数据库中的若干蛋白质形成训练集,若干蛋白质形成验证集,若干蛋白质形成测试集,其中训练集用于训练模型的训练;
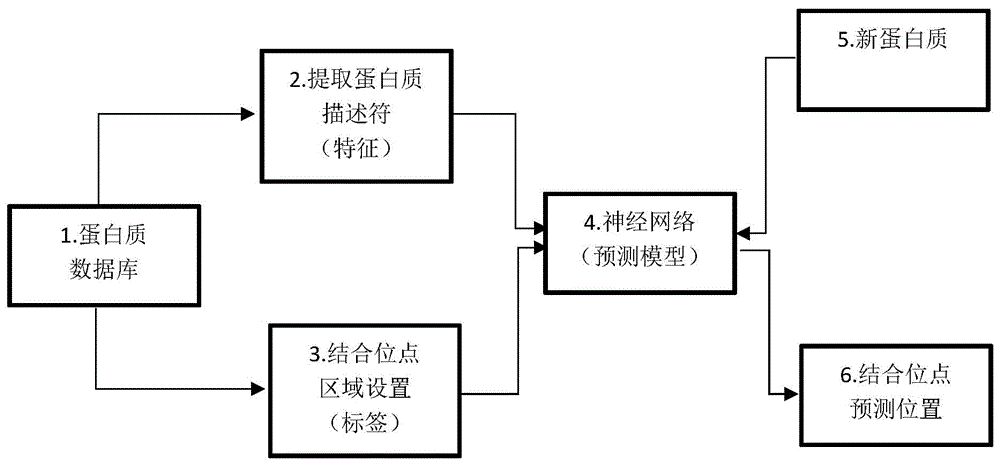
步骤2、训练后的训练模型对蛋白质数据库进行特征提取和标签提取,获取数据,完成神经网络的训练,得到预测模型;
步骤3、将新的蛋白质输入预测模型中,进行结合位点位置的定位和预测。
作为一种优选的实施方式,步骤1中蛋白数据库包括sc-pdb和pdbbind,其中sc-pdb中的蛋白质按照3:1:1的比例随机抽取蛋白质分别形成训练集、验证集和测试集,pdbbind中的所有蛋白质作为测试集。
作为一种优选的实施方式,步骤2中特征提取采用网格的多通道特征提取,对每个蛋白质进行计算,得到每个蛋白质的多通道网格。
作为一种优选的实施方式,网格的多通道特征提取的步骤如下:
步骤1、构建蛋白质的包围盒,并将蛋白质离散成
步骤2、设置原子网格通道;
步骤3、设置氨基酸网格通道;
步骤4、设置疏水性网格通道;
步骤5、设置能量通道网格;
步骤6、通过对每个蛋白质进行上述5个对蛋白质包围盒网格的处理步骤之后,得到每个蛋白质的4通道蛋白质网格值。
作为一种优选的实施方式,标签提取的步骤如下:
步骤1、将蛋白质的4通道网格按照步长5埃米进行
步骤2、设定每个取样块的标签,若取样块的中心点位于结合位点中心半径3埃米以内的范围内,则该取样块的标签设置为正样本,否则设置为负样本。
作为一种优选的实施方式,对单个蛋白质的负样本取样块按照其正样本取样块数量的2倍进行随机取样,最终使得每个蛋白质产生的取样块的正负样本比例为1:2。
作为一种优选的实施方式,将新的蛋白质输入预测模型后,首先进行对新的蛋白质进行取样,得到新的取样块,并对每个新的取样块进行结合位点概率预测,其次对新的取样块进行阈值筛选和聚类分析。
作为一种优选的实施方式,聚类分析包括对新的取样块进行聚类,在得到多个聚类类别之后,也就是多个结合位点之后,计算单个类别当中所有新的取样块的平均几何中心,将其视作该结合位点的最终中心。
作为一种优选的实施方式,阈值的设定值为0.5,聚类分析采用dbscan聚类算法。
采用了上述技术方案后,本发明的有益效果是:
1、能准确地预测结合位点的位置,由于采用了聚类分析,模型最终能预测和定位多个结合位点。
2、其综合考虑影响结合位点形成的因素,包括蛋白质的几何形状、氨基酸的疏水性以及分子间作用力等。
3、由于采用了神经网络构建模型,并且合理进行正负样本的划分,使得预测的结果的准确性得以保障。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明的整体流程图;
图2为蛋白质包围盒的构建以及网格化示意图;
图3为原子通道网格示意图;
图4为能量通道网格示意图;
图5为取样块的示意图;
图6为神经网络架构示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
根据图1至图6所示,一种基于深度学习的蛋白质药物结合位点预测方法,包括训练模型和预测模型,训练模型通过对已知的蛋白质数据库进行特征提取和标签提取,获取数据,完成神经网络的训练,得到预测用的模型,即预测模型,训练得到的模型对新的蛋白质进行相同的预处理后提取特征,进行结合位点位置的定位和预测。
蛋白质的数据库的选择将直接影响最终的预测效果。目前可用的蛋白质数据库包括pdb、sc-pdb、pdbbind等。其中,pdb数据库包含的种类最多,包括10多万条记录。但其数据条目错综复杂,甚至包括错误的数据。更为重要的是pdb数据库包含的物种千差万别,后期的筛选过程会非常复杂,因而作为药物预测的数据库并不合适。
sc-pdb是对pdb进行筛选之后得到的专门用于药物研究的数据库,并对蛋白质进行了预处理。sc-pdb包含蛋白质的结构文件、配体的结构文件以及结合位点的结构文件和位点信息等。其蛋白质、配体以及结合位点的结构文件都是采用mol2文件格式。该数据库对结合位点的描述非常详细,不仅包含结合位点的位置、三维结构文件和体积,还包含了位点的结合特性如极性表面积、疏水性以及b-factor等。基于sc-pdb的种种优点,本发明采用该数据库作为训练模型时的数据库。
pdbbind同样是一个描述蛋白质和配体结合的数据库。该数据库包含的数据不光包含蛋白质与配体,还包含蛋白质与核酸、蛋白质与蛋白质等结合信息。pdbbind的蛋白质采用pdb格式,而配体采用sdf和mol2格式。由于pdbbind也是描述蛋白质小分子结合的数据库,因此在本发明中作为进一步的验证数据库。
数据库中蛋白质样本需要经过特征提取作为神经网络的输入。本发明采用基于网格的多通道特征提取,即对每个蛋白质进行计算,得到每个蛋白质的多通道网格,详情如下:
如图2所示,对于每个样本蛋白质,首先,构建蛋白质的包围盒,并将蛋白质离散成
其次,如图3所示,设置原子网格通道。原子是蛋白质最基础的表现形式,也是构成蛋白质几何结构的最小单元,是蛋白质结构的载体,影响着蛋白质结合位点的形成。该通道的设置中,在得到的离散网格后,依次遍历蛋白质的原子,以该原子为中心,将该原子范德华半径范围内覆盖的网格的值设为该原子的值。原子的取值按照原子类型的不同依次设为整数。若不同原子的半径内覆盖到同一网格,则将该网格的值设为这些原子的平均值。经过以上处理后得到该蛋白质在几何结构通道上的网格值。
第三,设置氨基酸网格通道。氨基酸是构成蛋白质序列的基本单元,是蛋白质发挥功能的重要影响因素,处于不同位置的氨基酸残基可能带有结合位点的结合活性。在该通道的处理中,依次遍历该蛋白质的氨基酸,以每个氨基酸的几何中心为中心,将该中心4埃米范围内覆盖的网格的值设为该氨基酸残基的值。氨基酸的取值同样按照类型不同设为整数,多个氨基酸覆盖的网格值取平均值。这里取4埃米的原因是氨基酸的平均长度为8埃米,因此取4埃米作为半径。
第四,设置疏水性网格通道。疏水性是指分子对水的亲和性,是与水排斥程度的衡量标准,该属性是影响结合位点形成的一个重要因素。该通道同样遍历该蛋白质的氨基酸,将氨基酸中心4埃米覆盖的网格进行值的设置。不同的是,这里设置的值不再是氨基酸的类型值,而是氨基酸的疏水性数值。氨基酸的疏水性参考表如下,该表是以最疏水的苯丙氨酸设置为100、中性甘氨酸设置为0作对比并归一化之后的疏水性数值。
氨基酸疏水性数值参考表(ph=7)
最后,如图4所示,设置能量通道网格。范德华力是分子间普遍存在的作用力,是分子结合的关键因素,因而该因素也是影响结合位点形成的重要因素之一。得到蛋白质的包围盒离散网格后,依次将探针c原子放置在网格的各个位置,计算其与整个蛋白质的范德华力作用力作为该网格的值。最后得到c原子与蛋白质作用的范德华力作用力能量分布图,依次将c原子放置于网格的每个位置,计算其与蛋白质的范德华力作为该网格的值。
通过对每个蛋白质进行以上5个对蛋白质包围盒网格的处理步骤之后,得到每个蛋白质的4通道蛋白质网格值,用于之后的训练。
在训练之前,需要确定神经网络的输入以及正负样本。首先,因为蛋白质的大小并不相同,结合位点位置也不相同,每个蛋白质产生的4通道网格同样是长宽高各不一样的。如图5所示,本发明将蛋白质的4通道网格按照步长5埃米进行
在进行取样块的设定后,还需要设定每个取样块的标签。本发明首先确定结合位点的几何中心,若取样块的中心点位于结合位点中心半径3埃米以内的范围内,则将该取样块的标签设置为正样本,否则设置为负样本。当然,最终的正负样本会存在巨大的比例差。为确保正负样本足够均衡,对单个蛋白质的负样本取样块按照其正样本块数量的2倍进行随机取样,最终使得每个蛋白质产生的取样块的正负样本比例为1:2。
在确定好训练样本数量和标签之后,需要构建神经网络,本网络使用如图6所示。该神经网络模型的输出为每个取样块是结合位点的可能性,输出值在0-1之间。训练时正样本的标签值为1,负样本的标签值为0。
将sc-pdb中的蛋白质按照3:1:1的比例随机抽取蛋白质形成训练集、验证集和测试集。其中训练集用于训练模型的训练,验证集用于监测训练过程,选择何时停止训练模型。测试集用于最后验证模型效果。
在sc-pdb的测试集上完成测试后,为进一步确保模型的效果,还需要pdbbind上进一步验证模型效果,将pdbbind所有蛋白质设置成测试集,并进行测试。若该模型的测试成功率达到一定阈值,则该模型可以作为最终的预测模型。
训练之后得到的模型便可以进行预测。预测时,同样对新的未知蛋白质进行
本发明将已有的方法进行结合,提出一种能综合考虑各种影响结合位点形成因素的位点预测方法。一方面考虑了蛋白质的三维几何结构,另一方面,该发明将一些能量因素包括范德华力以及疏水性等考虑在内,充分结合发挥各方面的优势,避免各自的弊端,预测的位点的位置更加准确。
再者,本发明采用基于多通道网格的实现方式,网格的设置能充分将蛋白质原子进行映射。多通道的设置能将影响结合位点形成的各因素更加简单的表示成计算机中的形式。
最后,本发明采用基于深度学习的模型构建。深度学习能够拟合复杂的非线性映射,具有强大的表征能力,可以学习到更加丰富的特征。通过构建深度神经网络,将蛋白质的多通道网格作为输入进行训练,得到能够预测结合位点位置的智能预测模型。
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种尿道扩张器的制作方法
- 一种基于推杆电机的输液板的制...
- 棉签自动消毒装置的制作方法
- 一种心电异常检测方法、终端及...
- 健康状态管理系统、健康状态管...
- 临床决策支持的制作方法
- 用于识别色素沉着过度的斑点的...
- 用于分析心电图的用户界面的制...
- 信息处理装置、方法以及程序与...
- 基于云技术的电子处方传送系统...
- 还没有人留言评论。精彩留言会获得点赞!