
【论文标题】Protein language model embeddings for fast, accurate, alignment-free protein structure prediction
【作者团队】Konstantin Weissenow, Michael Heinzinger, Burkhard Rost
【发表时间】2021/08/02
【机 构】慕尼黑工业大学
【论文链接】https://www.biorxiv.org/content/10.1101/2021.07.31.454572v1.pdf
【代码链接】https://github.com/kWeissenow/ProtT5dst
【推荐理由】超大预训练模型Prottrans作者在结构预测方面新作
所有SOTA蛋白质结构预测都依赖于多序列比对(MSA)中捕获的进化信息,主要是进化耦合或共进化。这种信息并不对所有的蛋白质都可用,而且生成的计算成本也很高。基于人工智能的预测模型只使用单一序列作为输入,更容易、更便宜,但性能很差,速度变得无关紧要。在这里,我们描述了第一个有竞争力的人工智能解决方案,专门将从预训练的蛋白质语言模型中提取的嵌入,即从Transformer语言模型ProtT5中提取的嵌入,以单一序列输入到一个相对较浅的卷积神经网络中,该网络是根据残基间的距离,即二维的蛋白质结构训练的。主要的进步源于对ProtT5所学到的注意力头的处理。虽然没有达到SOTA,但我们的精简方法不需要任何MSA,在大大降低成本同时接近了依靠共进化的方法,从而加快了开发和每个后续预测的速度。通过产生特定的蛋白质而不是家族平均预测,这些新的解决方案可以区分具有类似结构的同家族蛋白质成员的结构特征。
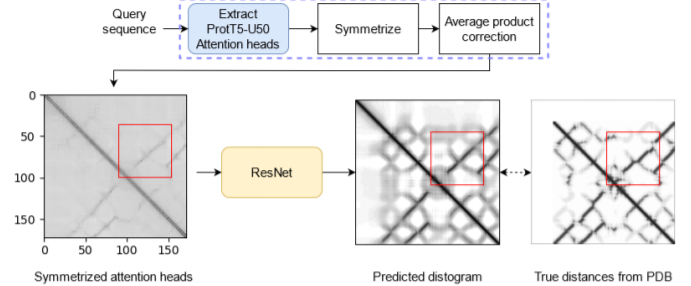
上图显示了本文的预测框架,例子显示了对CASP14中T1026应用对称性(APC,即average product correction )后的注意力头的平均值,表明ProtT5在无监督的情况下学习了蛋白质结构相关内容。残差网络(黄色)在不同的模型中是相似的,使用ProtT5、ProtBERT、ProtAlbert或SeqVec的一维蛋白质嵌入的模型调整了它们的结构。红色直线部分将蛋白质被分成64x64大小的区域,即64个连续残基。对于每块区域,CNN(ResNet)预测了相应的远距离接触图。比较用的MSA基线模型根据共进化的信息进行训练,通过生成MSA和估计Potts模型的参数,取代了蓝色虚线标记的模块。
最终我们使用pyRosetta的trRosetta折叠protocol的修改版来计算三维结构。与trRosetta不同的是,我们放弃了对角度信息的任何约束,使C-α距离而不是C-β距离被用作约束。我们首先使用我们预测的距离图中的短距离、中距离和长距离在不同的距离概率阈值水平(这里:[0.05, 0.5])作为约束条件生成了150个粗粒度的decoys,并通过使用pyRosetta的FastRelax protocol relax了50个模型。decoys的选择以及最终模型的选择是基于Rosetta的最低总能量。
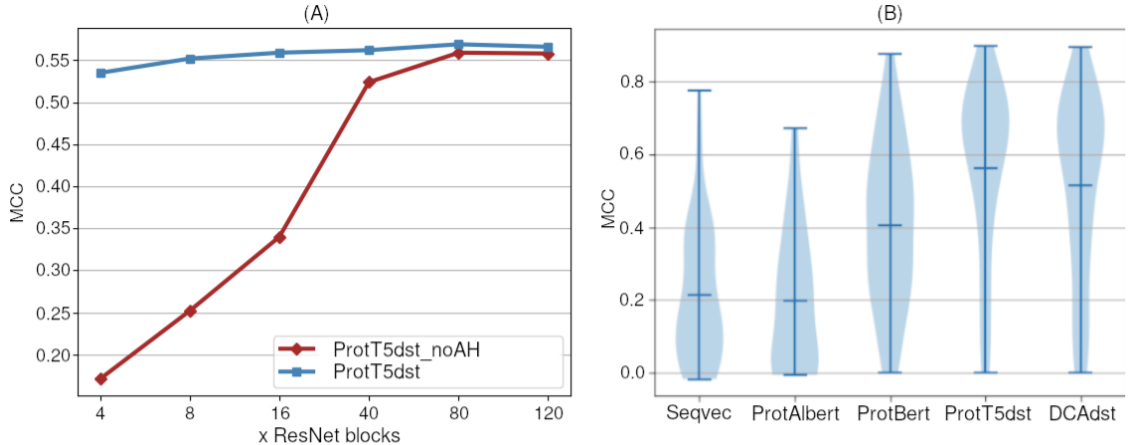
图A:SetValCASP12;X轴为ResNet块的数量;左侧描述了 "浅层CNN",即那些参数较少的CNN(从4块的235,306到120块的6,501,162)。两条线都使用了ProtT5 pLM:上面的蓝线是这里介绍的使用注意力头(AHs)的方法,下面的红线是没有AHs的嵌入。AHs已经在浅层架构中表现良好,而没有AHs的原始嵌入需要至少40个残差块才能达到0.5以上的MCC水平。
图B:SetValCASP12+SetTstCASP13:四种不同的pLMs的嵌入的小提琴图,以及使用共进化的方法(DCAdst)的结果,证明了即使没有学习MSA,ProtT5dst的预测效果优于共进化方法
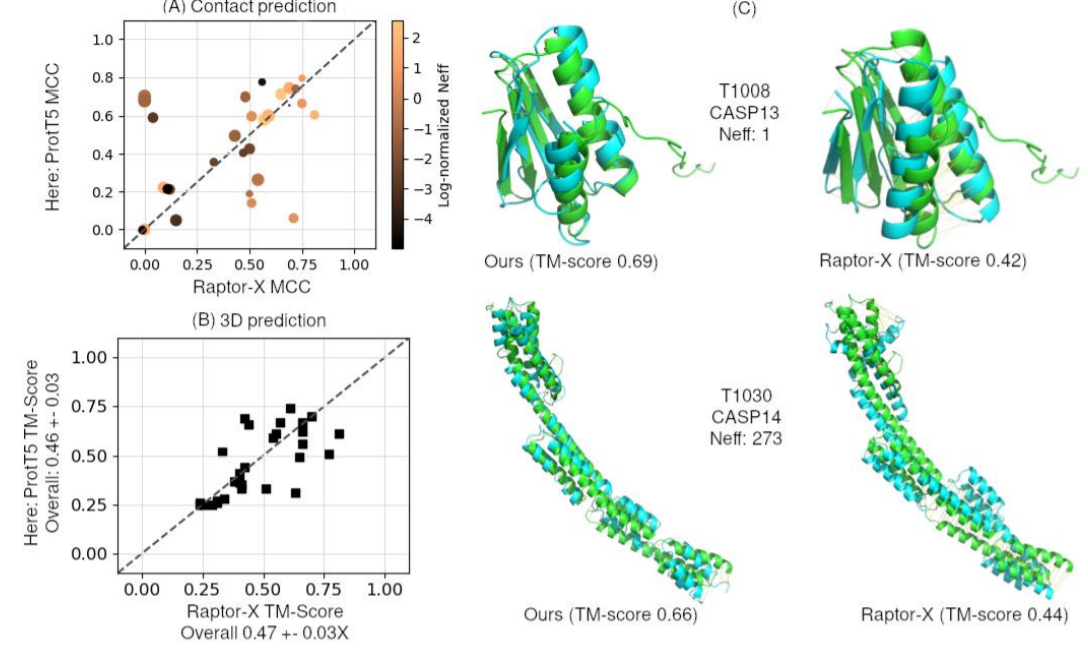
上图展示了ProtT5dst与RaptorX的对比。数据集为SetTstCASP13 + SetTstCASP14,方法为ProtT5dst以及Raptor-X。
图A:中距离和长距离接触预测的MCCs的比较。
图B:三维结构预测性能,TMalign计算了所有预测的TM-分数。总体而言,两种方法的性能相似。
图C:两个蛋白质(T1008和T1030;实验:绿色,预测:青色)的三维预测与实验的详细比较。一个蛋白质较小的(T1008),另一个较大(T1030),尽管这两种蛋白质的总体表现相似,但ProtT5的表现都优于Raptor-X。对于T1008(CASP13),两种预测都正确地捕获了整体折叠,但Raptor-X错误地调换了两个螺旋,使TM分数从0.69(ProtT5dst)降低到0.42。同样,对于较长的蛋白质T1030(CASP14),Raptor-X错置了几个螺旋。
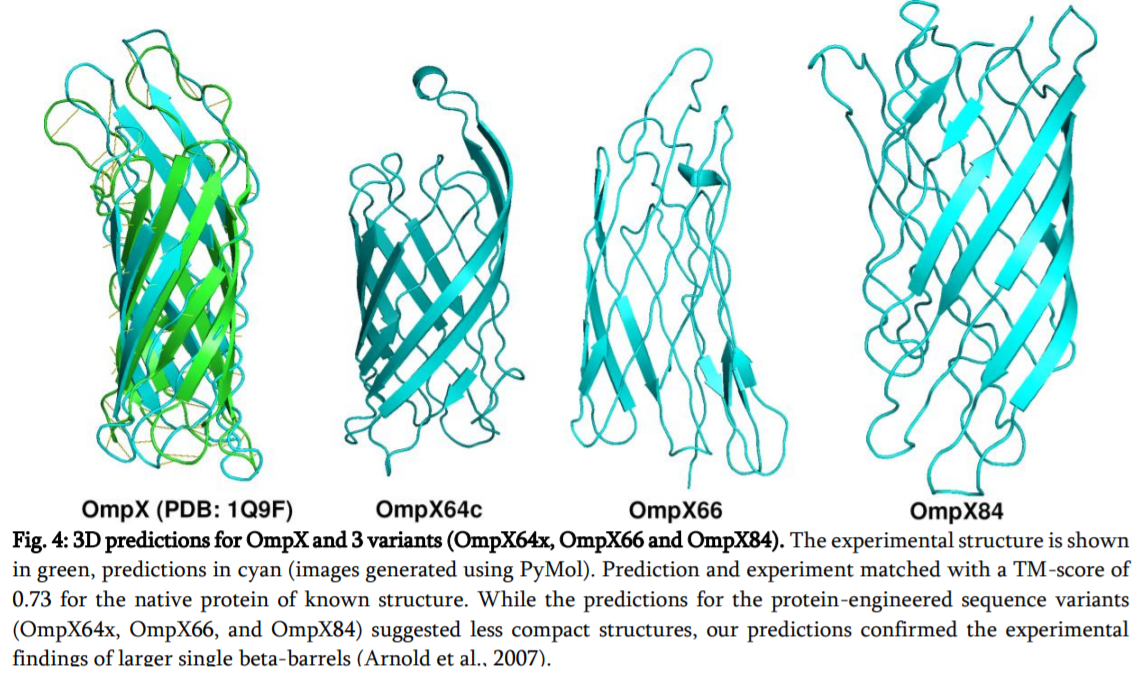
上图展示了OmpX和3个变体(OmpX64x, OmpX66和OmpX84)的三维预测。实验结构显示为绿色,预测为青色。对已知结构的本地蛋白,预测和实验相匹配的TM分数为0.73。虽然对蛋白工程序列变体的预测表明结构不太紧凑,但我们的预测证实了较大的单β-barrels的实验结果。
- 结论
我们表明,基于来自单一蛋白质序列的嵌入的二维间距离预测在最近几年有了明显的改善,现在可以与共进化方法的性能相媲美。虽然我们的方法还没有提高SOTA的性能,但在不牺牲预测精度的情况下,推理时间的大幅减少提供了一个关键的实际优势。
更重要的是,我们的方法首次提供了基于单个蛋白质序列的准确的蛋白质结构预测,这对依赖不同MSA的以家族为中心的方法来说是有竞争力的。由于结构预测可以在几秒钟内获得,我们的方法可以很容易地为蛋白质结构预测的高通量分析提供基础,如虚拟结构突变。使用嵌入和共进化信息的方法很可能在未来共存互补。在未来,我们将通过预测结构差异来过滤比对的序列研究使用我们的方法进行MSA refinement的可行性。

评论
沙发等你来抢