蛋白对接_RosettaDock: 蛋白-蛋白复合物对接预测
最新推荐文章于 2024-04-18 09:21:16 发布

 好爱赵辰龙
好爱赵辰龙
 最新推荐文章于 2024-04-18 09:21:16 发布
最新推荐文章于 2024-04-18 09:21:16 发布
 阅读量3.6k
阅读量3.6k
 收藏
11
收藏
11


 点赞数
1
点赞数
1
作者: 吴炜坤
一文基本搞懂RosettaDock....
参考1: https://www. rosettacommons.org/demo s/latest/tutorials/Protein-Protein-Docking/Protein-Protein-Docking
参考2: Protein–Protein Docking with Backbone Flexibility
参考3: Conformer Selection and Induced Fit in Flexible Backbone Protein–Protein Docking Using Computational and NMR Ensembles
参考4: High-resolution protein–protein docking
参考5: Benchmarking and Analysis of Protein Docking Performance in Rosetta v3.2
一、前言
蛋白-蛋白对接是一个既复杂,又重要的话题。从一开始的基于FFTs算法的刚性对接ZDOCK发展到现在整合多步骤的HADDOCK、ClusPro、SwamDock等等,该领域的算法不断地升级迭代。
RosettaDock是蛋白-蛋白对接领域的一名老将,久经CAPRI的考验。特别擅长于蛋白-蛋白的局部构象的探索。包括已经发表的许多文章都是使用RosettaDock进行最后的优化,其地位可见一斑。
近年来,RosettaDock也发展出了针对特殊蛋白家族提供的算法如针对抗体-抗原对接的SnugDock、同源多聚体组装对接的SymmetricDock、以及多肽-蛋白对接的FlexPepDock、以及为高难度对接任务开发的Motif Score的RosettaDock4.0等等。
本文将简要的介绍常规的RosettaDock3.2蛋白-蛋白对接的基本框架和用法。
二、基本原理和算法
RosettaDock的基本算法如下:
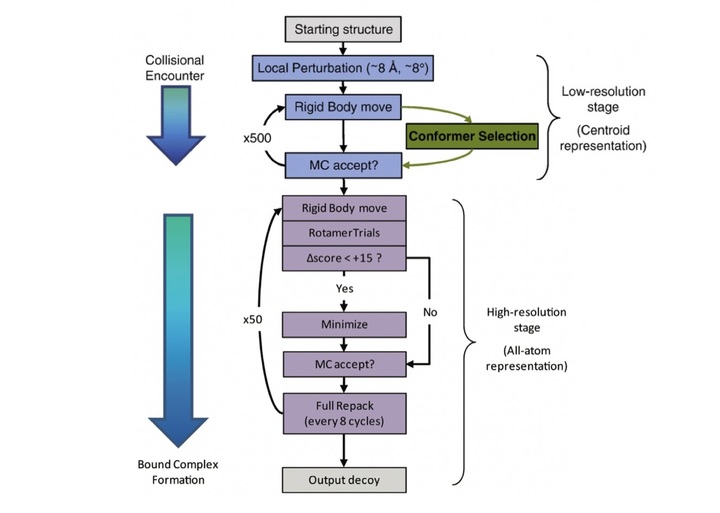
整个对接流程分为两大部分,在第一个低分辨率阶段,蛋白质之间的侧链构象被一个粗粒化球所替代。直接搜索蛋白质之间的骨架形状相适配的程度。在第二个阶段才会考虑全部的侧链构象,计算更加精确的相互作用能量。
起始的局部干扰,将初猜构象的其中一个组分随机平移和转动8埃和8°(或8埃,3°)。
在低精度阶段,进行500次刚性体的移动,其中可以选择是否进行Ensemble的构象交换选择。输出最低的构象,进行高精度阶段
在高精度阶段,进行50次MCMPCycle:
- repack构象并能量最小化,作为初始出发构象。
- 内部循环: 运行50次MCMCycle, 每次MCMCycle包括: 刚性体移动、RotamerTrials优化每一个氨基酸为最低能量状态、并判断能量是否大于15REU,如果能量下降太小,运行一次刚性体之间的能量最小化。(每8步MCMCycle运行一次Repack(repack模式可以分为rt_min或sc_min))
- 外部循环: 重置初始状态,并运
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 1万+
1万+










 暂无认证
暂无认证





 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









