基因表达数据的聚类分析方法

 最新推荐文章于 2023-11-18 15:56:39 发布
最新推荐文章于 2023-11-18 15:56:39 发布
 阅读量9.7k
阅读量9.7k
 收藏
35
收藏
35


 点赞数
8
点赞数
8
介绍
基因表达(gene expression) 是指将来自基因的遗传信息合成功能性基因产物的过程。
基因表达产物通常是蛋白质,但是非蛋白质编码基因如转移RNA(tRNA)或小核RNA(snRNA)基因的表达产物是功能性RNA。
所有已知的生命,无论是真核生物(包括多细胞生物)、原核生物(细菌和古细菌)或病毒,都利用基因表达来合成生命的大分子。
基因编码并可用于合成蛋白质,这个过程称为基因表达。
在像人类这样的高等生物中,根据细胞类型(神经细胞或心脏细胞)、环境和疾病状况等各种因素,数以千计的基因以不同的量一起表达。
例如,不同类型的癌症在人类中引起不同的基因表达模式。可以使用微阵列( Microarray )技术研究不同条件下的这些不同基因的表达模式。
微阵列和基因表达谱
来自微阵列的数据可以想象为矩阵或网格,矩阵中的每个单元格对应于特定条件下的基因表达值。
如下图所示,矩阵的每一行对应一个基因 gi ,每一列对应一个条件/样本 si

人类有大约 20,000 个表达基因,假设我们想知道它们的表达模式,即在不同类型的人类癌症下哪些基因产生更高或更低水平的蛋白质。
另外,假设已知有 20 种人类癌症,那么微阵列基因表达矩阵的结果就有 20,000 行对应基因,20 列对应于 20 种癌症。
基因表达聚类
分析基因表达数据的第一步是在经典数据挖掘中对基因或样本进行聚类。
可以根据基因在所有条件下的表达模式对基因进行聚类,并且可以使用所有基因的基因表达模式对样本进行聚类。
关于聚类问题
对于基因聚类,数据点是基因,特征是所有样本的表达值。
因此,在针对癌症示例的基因聚类中,将聚类 20,000 个数据点( data-points ),每个点具有 20 个维度。
聚类基因表达数据提供了对基因共调控(co-regulation)和基因细胞功能的重要见解。
聚集在一起的基因在所有样本中具有相似的表达模式,这可能表明这些基因的共同调控。
此外,来自同一簇的基因可能执行类似的细胞功能,这有助于注释新发现的基因。
相反,对于样本聚类,样本是使用跨所有基因的基因表达量作为特征进行聚类的数据点。由此将聚类 20 个数据点,每个点具有 20,000 个维度。
下面,我们将讨论执行聚类的不同方法
- Llyod’s
- K-均值聚类 ( K-means clustering )
- 层次聚类 ( Hierarchical Clustering )
邻近计算
邻近计算(Proximity calculation)
用于聚类的数据点之间的距离或接近度很重要,因为所有聚类算法的工作原理都是将近点聚集在一个聚类中。
使用 Pearson 相关系数中的特征计算数据点 Oi 和 Oj 之间距离的有效措施之一:
Pearson( O i O_i Oi, O j O_j Oj) = ∑ d = 1 p ( O i d − μ o i ) ( O j d − μ o j ) ∑ d = 1 p ( O i d − μ o i ) 2 ∑ d = 1 p ( O j d − μ o j 2 ) \frac {\sum_{d=1}^p(O_{id} - \mu_{oi} )(O_{jd} - \mu_{oj} )}{\sqrt {\sum_{d=1}^p(O_{id} - \mu_{oi})^2} \sqrt {\sum^p_{d=1}(O_{jd} - \mu_{oj}^2)} } ∑d=1p(Oid−μoi)2∑d=1p(Ojd−μoj2)∑d=1p(Oid−μoi)(Ojd−μoj)
K均值聚类
k均值聚类算法(k-means clustering algorithm)
是一种迭代求解的聚类分析算法。属于无监督学习算法。
步骤:
预将数据分为K组,则随机选取K个对象作为初始的聚类中心,然后计算每个对象与各个种子聚类中心之间的距离,把每个对象分配给距离它最近的聚类中心。
聚类中心以及分配给它们的对象就代表一个聚类。每分配一个样本,聚类的聚类中心会根据聚类中现有的对象被重新计算。这个过程将不断重复直到满足某个终止条件。
以下是一个二维数据。通过查看散点图,数据似乎包含 3 个不同的聚类。
因此,我们将任意发起 3 个聚类质心(cluster centroids)或聚类中心(cluster centers)。由于我们还没有任何聚类,这些质心(centroids)是空间中的任意点。
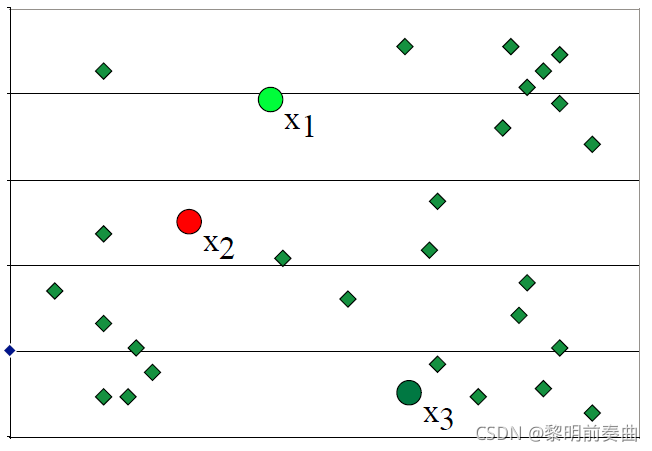
然后,我们计算所有点与 3 个质心的距离,并将这些点分配到它们最近的聚类。然后,我们使用聚类中分配的点重新计算质心。
聚类中心只是聚类中所有点的平均值。
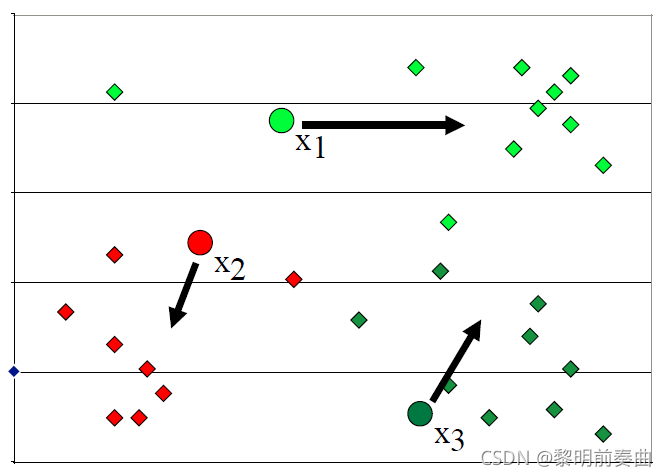
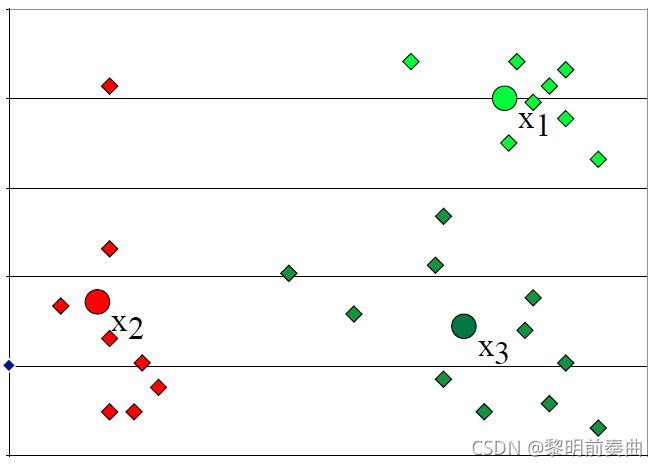
重新计算点与 3 个新分配的质心的距离,并将这些点重新分配到它们最近的聚类。
在点被重新分配到它们最近的聚类后,重新计算聚类中心。

重复上述步骤直到中心点收敛(convergence),基本上不在发生变化或满足精度为止。
层次聚类
层次聚类(Hierarchical Clustering)
是一种渐进式聚类技术,它从小簇开始,逐渐将密切相关的小簇合并成更大的簇,直到只剩下一个大簇为止。
相对于 K-means 的最大优势之一是层次聚类不必预先定义聚类的数量。相反,可以在聚类过程完成后推断最佳聚类数。
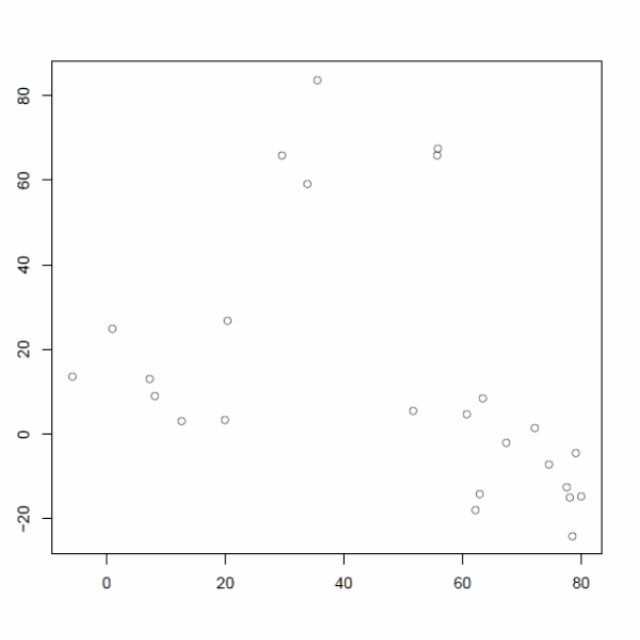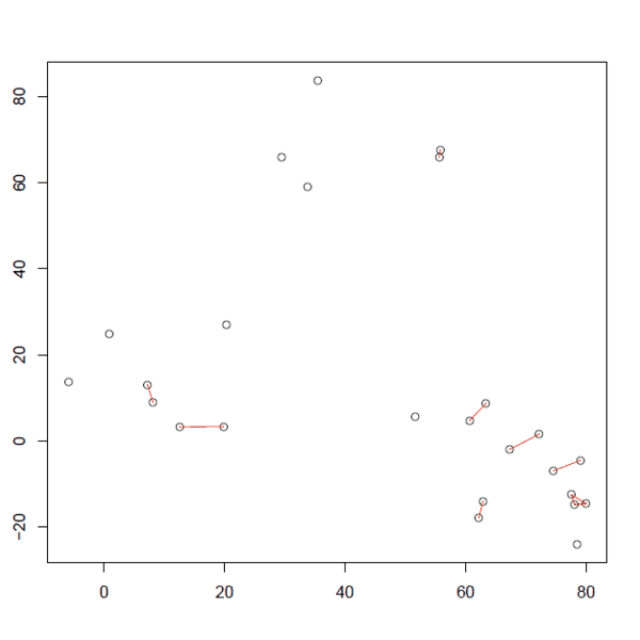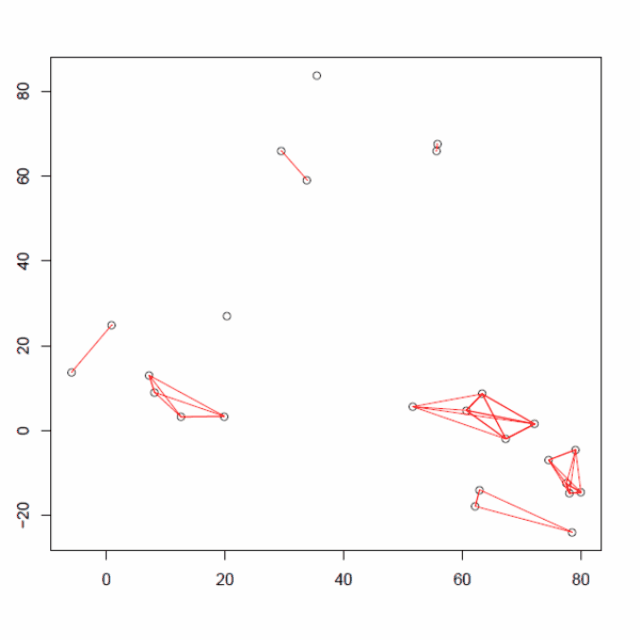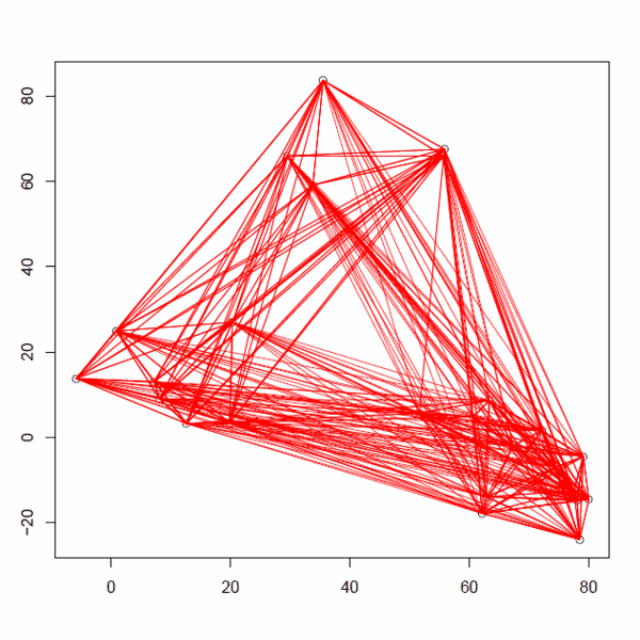
使用以下包含 25 个数据点的二维数据仔细研究层次聚类算法
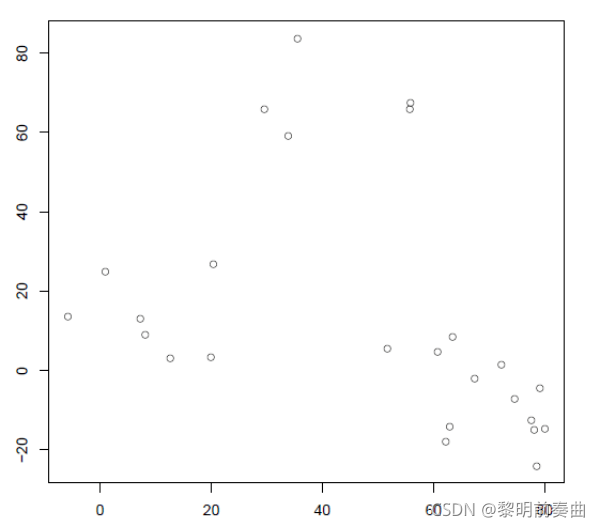
-
将每个点分配给它自己的单个簇,即有 25 个簇,每个簇包含 1 个点。
-
然后,计算每个聚类中心点。
-
计算所有的质心距离并将两个簇连接到一个质心最近的新簇中。重新计算新形成的簇的质心。
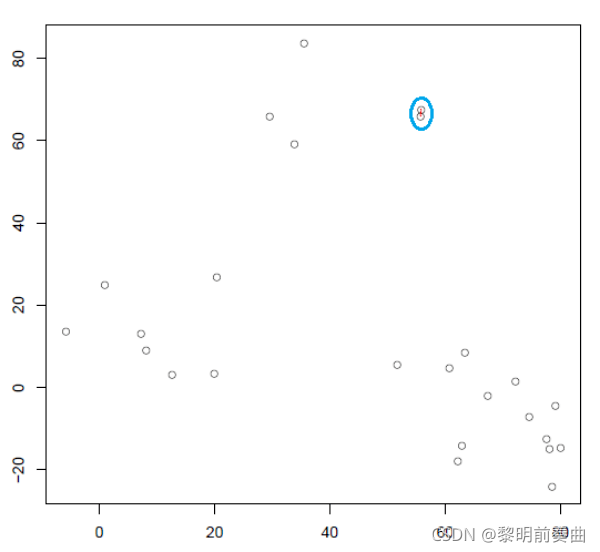
迭代 1
再次计算所有的质心距离,并检测最近的两个簇并将其连接到一个新簇中。重新计算新簇的质心。
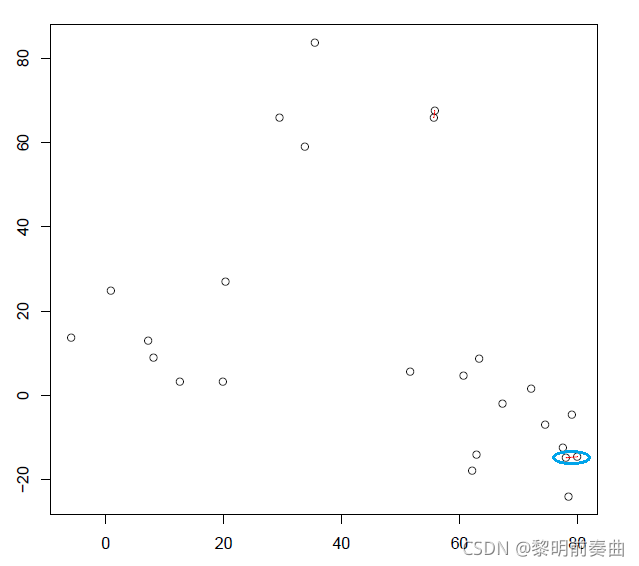
迭代 2
重复3个步骤,计算所有的质心距离,合并2个最近的簇,重新计算新形成的簇的质心,直到只得到一个包含所有25个数据点的大簇(收敛)。

动图展示
整个层次聚类过程可以使用如下所示的树状图进行可视化,其中分叉树的叶节点是数据点,内部节点显示执行的每个合并步骤。
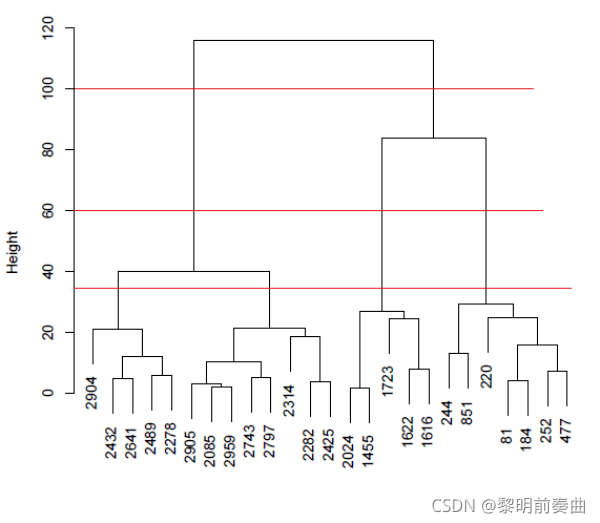
左侧的高度比例显示了聚类合并的距离
最低的内部节点距离很小,表明最近的簇或点首先被合并。
最高的内部节点距离很远,表示相距很远的点或簇以最高距离连接到一个簇中。
实际的聚类解决方案是通过在指定距离截止点处跨聚类树状图绘制一条水平线来获得的。
簇数等于水平切割线遇到的交点数。
例如,在距离截止值(distance cutoff)=60 处绘制的红色水平线为 25 个数据点定义了 3 个clusters。
例子
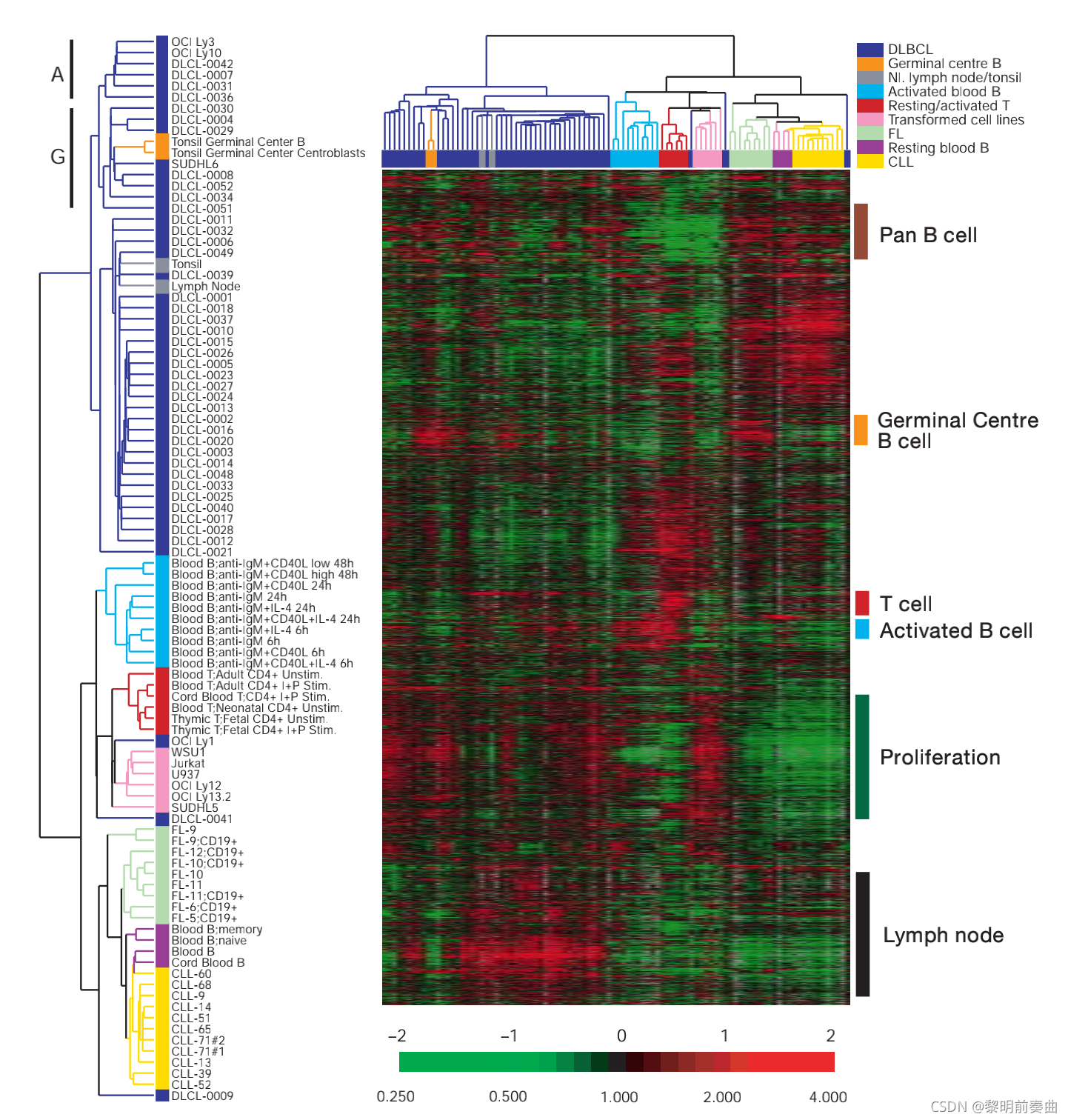
一个例子显示了通过基因表达数据的层次聚类识别的不同类型的弥漫型B大细胞淋巴瘤(diffuse large B-cell lymphoma)。
根据确定的不同类型,我们对癌症预期如何发展的估计会有所不同,并且还可能导致处方治疗的差异。














 1万+
1万+










 暂无认证
暂无认证




















 到【灌水乐园】发言
到【灌水乐园】发言










peacezha: -l <INT=3> 清除重复的级别。0表示禁用,1表示仅清除包含的单倍体,2表示清除所有类型的单倍体,3表示以最积极的方式清除所有类型的单倍体。默认情况下,3表示非trio装配,0表示trio装箱装配。对于trio装箱装配,仅允许级别0和级别1,可以看看这一篇https://blog.csdn.net/weixin_45898964/article/details/138682488
peacezha: 可以看看这一篇https://blog.csdn.net/weixin_45898964/article/details/138633726?spm=1001.2014.3001.5501
weixin_51083352: putty里的conda怎么安装
Yorleleiyo: 引用「tab分割的基因对」 tab分割的基因对是什么意思?
2301_77522275: 能教一教那些依赖如何安装嘛